Synthesizability, hardness, and stacking order in multicomponent transition metal carbides from machine-learned potentials
Pith reviewed 2026-06-29 06:49 UTC · model grok-4.3
The pith
Multicomponent carbides mixing group 4/5 and group 6 metals form stacking-ordered phases with formation energies below those of disordered rocksalt or hexagonal structures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
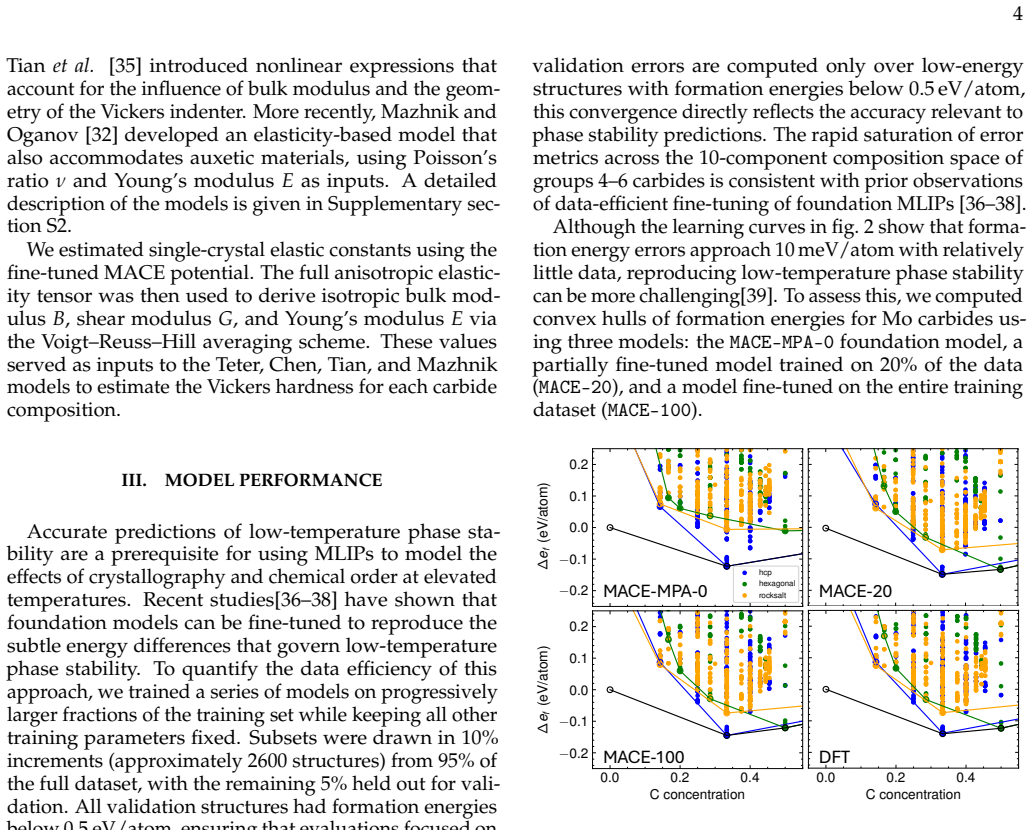
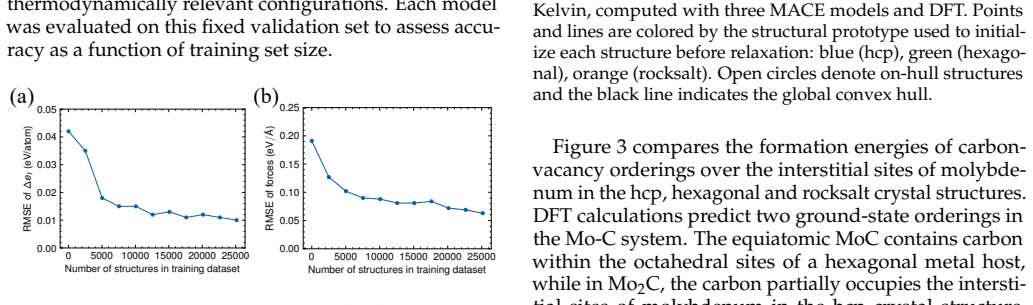
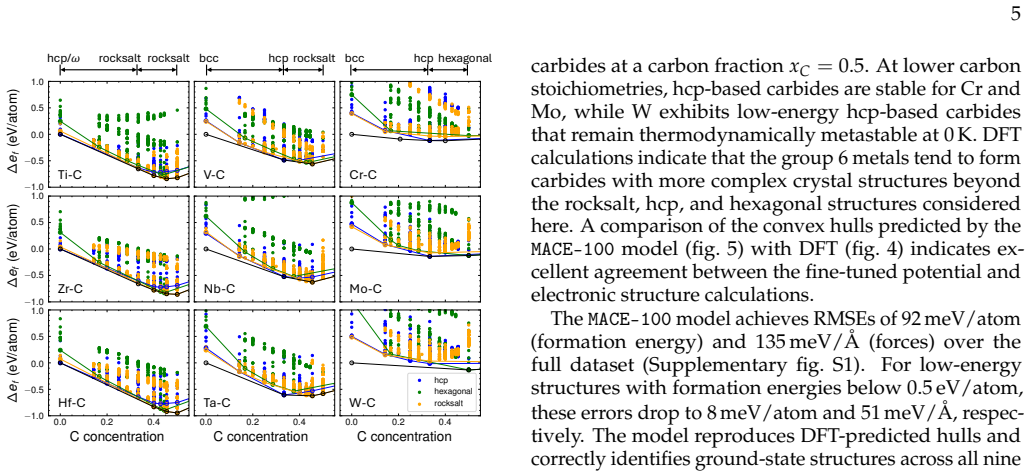
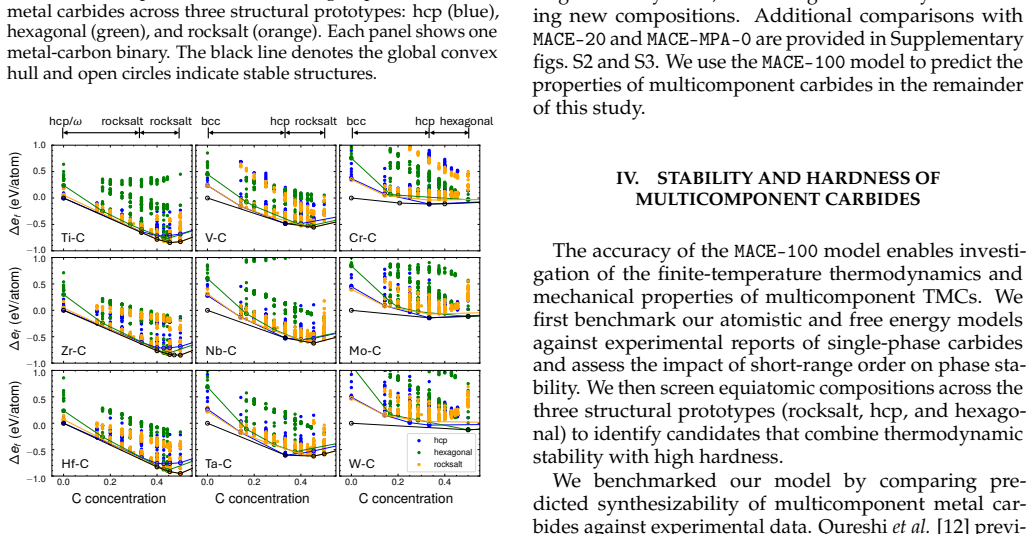
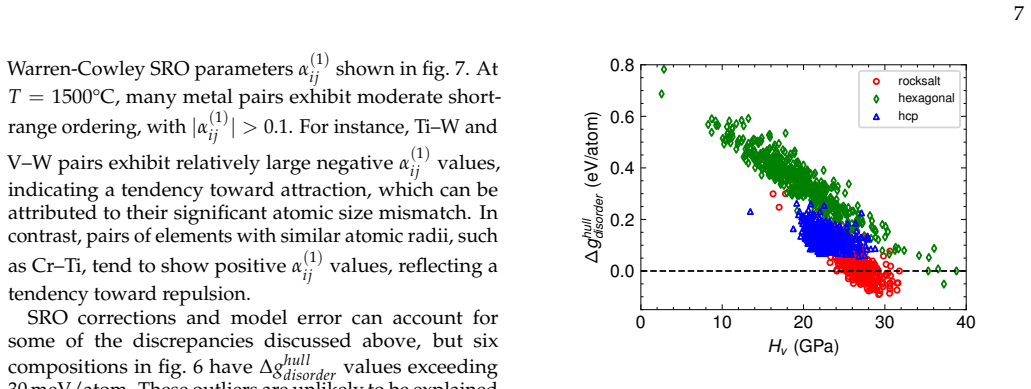
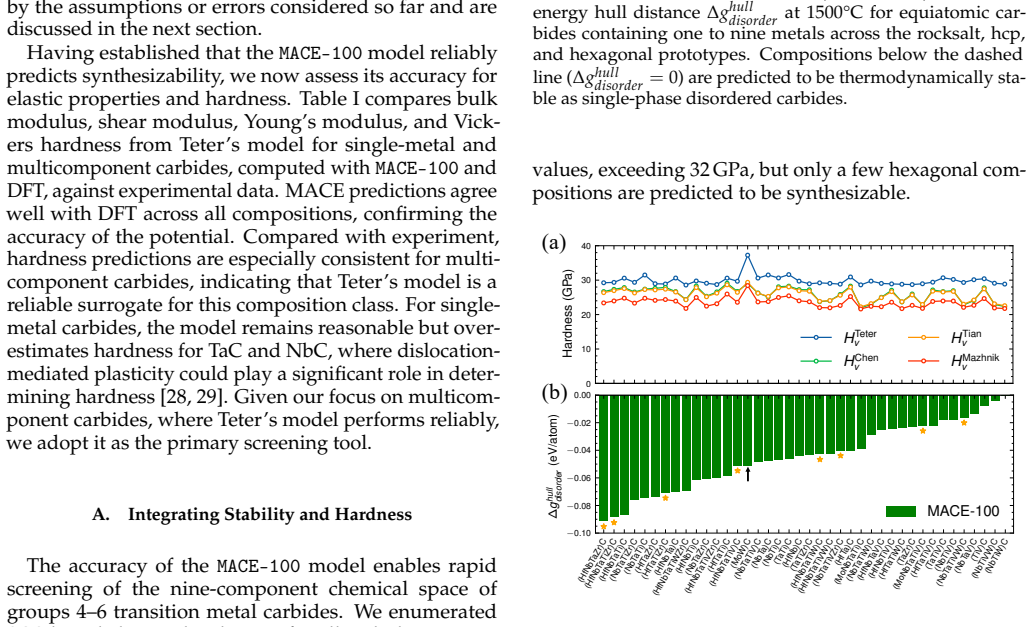
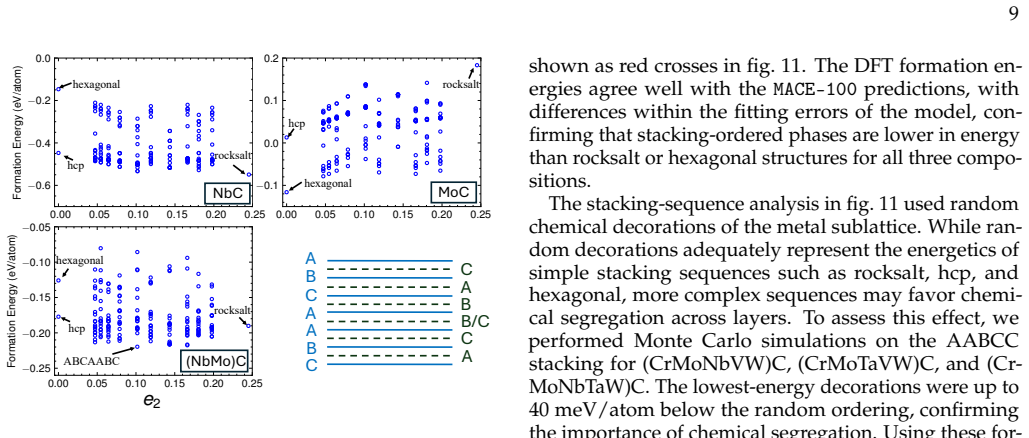
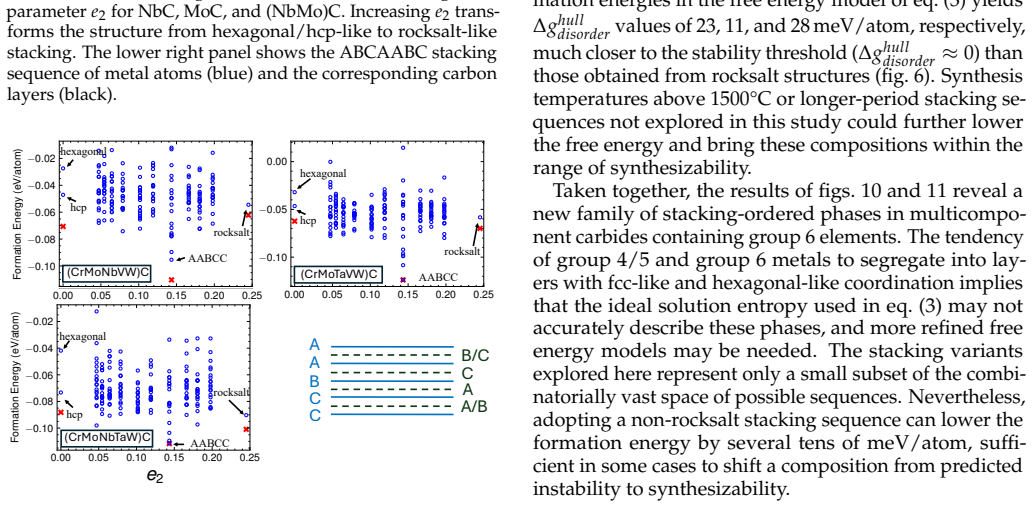
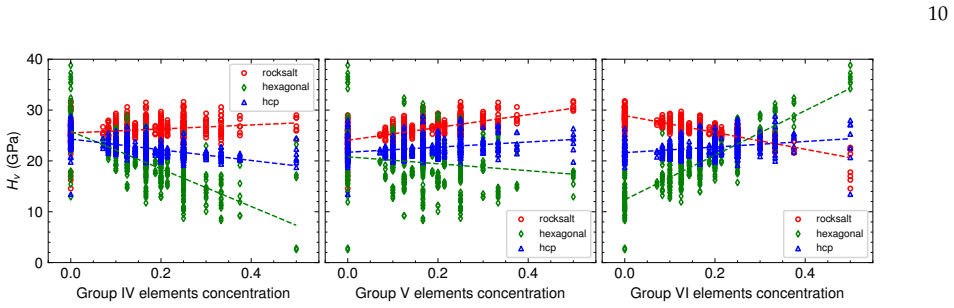
Fine-tuning the MACE potential on roughly 28,000 density functional theory calculations enables accurate prediction of formation energies to about 10 meV per atom across the nine-component space. Screening more than 1500 compositions shows that mixing group 4/5 and group 6 metals stabilizes a previously unreported class of stacking-ordered carbide phases whose energies fall well below those of disordered rocksalt and hexagonal structures. Density functional theory calculations on the predicted structures confirm the energy ordering and suggest experimental accessibility at 1500 °C.
What carries the argument
The fine-tuned MACE machine-learned interatomic potential, which ranks formation energies and elastic properties for rapid screening of multicomponent compositions and identifies the stabilizing effect of stacking order in mixed-group carbides.
If this is right
- Group number of the metals controls both thermodynamic stability and hardness across the full composition space.
- Free-energy contributions from short-range order are only a few meV per atom, validating the disordered solid-solution approximation for high-throughput searches.
- Synthesizability predictions at 1500 °C match known experimental single-phase and multiphase carbide behavior.
- Stacking-ordered phases constitute a distinct, lower-energy family accessible only through multicomponent mixing.
Where Pith is reading between the lines
- Targeted synthesis experiments could focus on equiatomic mixtures that combine early and late transition metals to test the predicted stacking order.
- The same screening workflow could be applied to other multicomponent ceramics where stacking order might similarly lower energy.
- Elastic-property predictions suggest these phases may offer hardness advantages that warrant direct nanoindentation measurements once synthesized.
Load-bearing premise
The fine-tuned potential remains accurate enough to rank the thermodynamic stability of the new stacking-ordered structures correctly even when trained on only 20 percent of the density functional theory data.
What would settle it
Synthesis of one predicted stacking-ordered multicomponent carbide (for example, a Ti-Zr-Mo-C composition) at 1500 °C followed by X-ray diffraction that either confirms or rules out the ordered stacking sequence.
Figures

read the original abstract
Multicomponent transition metal carbides are promising for extreme-environment applications, but identifying compositions that are both synthesizable and hard remains challenging. We fine-tune the MACE machine-learned interatomic potential on approximately 28,000 density functional theory calculations spanning the composition space of groups 4-6 transition metals and carbon to predict the thermodynamic stability and elastic properties of multicomponent carbides. The fine-tuned model achieves formation energy errors of ~ 10 meV/atom for thermodynamically relevant structures with only 20% of the training data. We screen over 1500 equiatomic compositions across rocksalt, hexagonal, and hcp prototypes, combining free energy models with elasticity-based hardness surrogates. Synthesizability predictions at 1500{\deg}C agree well with experimental reports for both single-phase and multiphase carbides. The group number of the constituent metals governs both stability and hardness. Free energy contributions from short-range order are small, typically a few meV/atom, indicating that a perfectly disordered solid solution provides a reasonable approximation for high-throughput screening. For compositions mixing group 4/5 and group 6 metals, we identify a new family of stacking-ordered phases with formation energies well below those of disordered rocksalt or hexagonal structures. DFT calculations corroborate these predictions and suggest that stacking-ordered phases should be experimentally accessible in multicomponent carbides. This study provides a framework for screening synthesizable multicomponent materials with target properties, identifies promising carbide compositions across the full nine-component space, and reveals a new class of stacking-ordered carbides accessible only in multicomponent compositions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
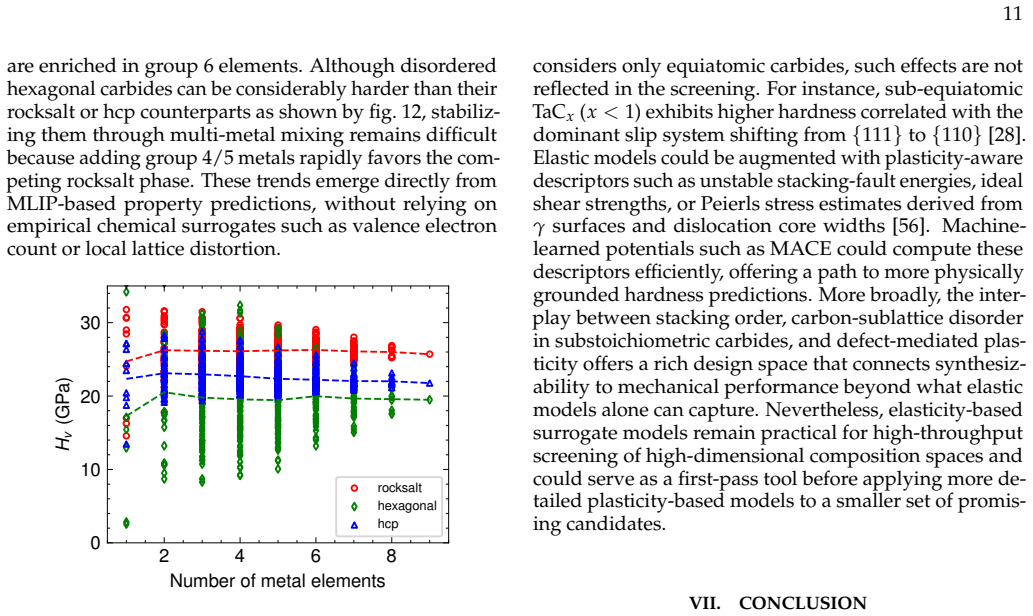
Summary. The manuscript claims that fine-tuning the MACE ML interatomic potential on ~28k DFT calculations for groups 4-6 transition metal carbides enables high-throughput screening of >1500 equiatomic compositions for thermodynamic stability and elastic hardness. It reports that metal group number governs both properties, that short-range order free-energy contributions are small (~few meV/atom), that synthesizability predictions at 1500°C match experiments, and that a new family of stacking-ordered phases in group 4/5 + group 6 mixtures has formation energies well below those of disordered rocksalt or hexagonal structures; DFT calculations on these phases are said to corroborate the ML predictions and indicate experimental accessibility.
Significance. If the central claims hold, the work supplies a practical ML-accelerated framework for screening synthesizable multicomponent carbides with target mechanical properties across a nine-element space, demonstrates that group number is a dominant descriptor, and identifies a previously unreported class of stacking-ordered carbides whose stability appears accessible only in multicomponent compositions. The agreement between ML-based synthesizability predictions and existing experiments, together with the explicit DFT corroboration step for the new phases, strengthens the practical utility of the approach.
major comments (2)
- [Abstract] Abstract and methods: the reported average formation-energy error of ~10 meV/atom after training on only 20% of the DFT set is presented without (i) a breakdown of the train/validation split, (ii) error statistics specifically on stacking-ordered or long-period structures, or (iii) propagation of this uncertainty into the stability ranking that selects the new family; because the central discovery rests on ML-driven identification of phases whose energies are claimed to lie “well below” disordered prototypes, this omission is load-bearing.
- [Results] Results on new phases: while DFT is stated to corroborate the stacking-ordered predictions, the manuscript does not report how many candidate compositions were advanced from the 1500-composition ML screen to DFT, nor the magnitude of the formation-energy differences relative to the 10 meV/atom uncertainty; if those differences fall inside or near the model error, the claim that the new family is distinctly more stable (and therefore experimentally accessible) requires additional targeted validation.
minor comments (2)
- [Methods] The description of the elasticity-based hardness surrogate and its calibration against known carbides should be expanded for reproducibility.
- [Figure captions] Figure captions and text should explicitly state the temperature and reference states used for the free-energy comparisons that underpin the synthesizability predictions.
Simulated Author's Rebuttal
We thank the referee for their thorough review and valuable feedback on our manuscript. The comments raise important points regarding the validation of our machine learning model and the robustness of our findings on the new stacking-ordered phases. We will revise the manuscript to incorporate the requested details and clarifications.
read point-by-point responses
-
Referee: [Abstract] Abstract and methods: the reported average formation-energy error of ~10 meV/atom after training on only 20% of the DFT set is presented without (i) a breakdown of the train/validation split, (ii) error statistics specifically on stacking-ordered or long-period structures, or (iii) propagation of this uncertainty into the stability ranking that selects the new family; because the central discovery rests on ML-driven identification of phases whose energies are claimed to lie “well below” disordered prototypes, this omission is load-bearing.
Authors: We agree with the referee that these details are important for assessing the reliability of our predictions. In the revised manuscript, we will expand the Methods section to include a full breakdown of the train/validation split used in fine-tuning the MACE potential. We will also report error statistics broken down by structure type, including specifically for stacking-ordered and long-period structures. Furthermore, we will add an analysis in the Results section that propagates the model uncertainty into the stability rankings, demonstrating that the energy differences for the identified stacking-ordered phases remain significant relative to the ~10 meV/atom error. These revisions will directly address the load-bearing nature of this information for our central claims. revision: yes
-
Referee: [Results] Results on new phases: while DFT is stated to corroborate the stacking-ordered predictions, the manuscript does not report how many candidate compositions were advanced from the 1500-composition ML screen to DFT, nor the magnitude of the formation-energy differences relative to the 10 meV/atom uncertainty; if those differences fall inside or near the model error, the claim that the new family is distinctly more stable (and therefore experimentally accessible) requires additional targeted validation.
Authors: We acknowledge that the manuscript would benefit from more explicit reporting on the DFT validation step. In the revision, we will specify the number of candidate compositions (selected based on ML-predicted stability) that were advanced to full DFT calculations for corroboration. We will also provide the quantitative formation-energy differences between the stacking-ordered phases and the disordered rocksalt/hexagonal structures, along with a comparison to the model uncertainty. This will include showing that the differences are well outside the error margin, supporting the claim of distinct stability and experimental accessibility. We believe this additional information will strengthen the presentation without altering the conclusions. revision: yes
Circularity Check
No significant circularity; ML screening validated by independent DFT
full rationale
The derivation uses a MACE potential fine-tuned on external DFT data (~28k calculations) to screen 1500 compositions and flag new stacking-ordered phases. The paper then performs separate DFT calculations that corroborate the ML predictions for those phases. No step reduces a claimed prediction to a fitted input by construction, no self-citation chain carries the central claim, and no ansatz or uniqueness result is smuggled in. The ~10 meV/atom error is an accuracy statement, not a definitional loop. The workflow therefore remains externally grounded.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The fine-tuned MACE potential achieves formation energy errors of ~10 meV/atom sufficient for reliable thermodynamic stability ranking across the screened composition space
- domain assumption Free energy contributions from short-range order are small (a few meV/atom) and therefore a perfectly disordered solid solution is a reasonable approximation for high-throughput screening
Reference graph
Works this paper leans on
-
[1]
B. C. Wyatt, S. K. Nemani, G. E. Hilmas, E. J. Opila, and B. Anasori, Ultra-high temperature ceramics for extreme environments, Nature Reviews Materials9, 773 (2024)
2024
-
[2]
W. S. Williams, Transition-metal carbides, Progress in Solid State Chemistry6, 57 (1971)
1971
-
[3]
F. Wang, X. Zhang, X. Yan, Y. Lu, M. Nastasi, Y. Chen, and B. Cui, The effect of submicron grain size on thermal sta- bility and mechanical properties of high-entropy carbide ceramics, Journal of the American Ceramic Society103, 4463 (2020)
2020
-
[4]
Zhang and M
R.-Z. Zhang and M. J. Reece, Review of high entropy ce- ramics: design, synthesis, structure and properties, Journal of Materials Chemistry A7, 22148 (2019)
2019
-
[5]
Prats and M
H. Prats and M. Stamatakis, Transition Metal Carbides as Supports for Catalytic Metal Particles: Recent Progress and Opportunities, The Journal of Physical Chemistry Letters 15, 3450 (2024)
2024
-
[6]
X. Bie, B. Ren, X. Zhou, S. Brahimi, S. Yue, and J. Song, Hy- drogen trapping and diffusion in sub-stoichiometric vana- dium and niobium carbide precipitates in high-strength steels, International Journal of Hydrogen Energy203, 153140 (2026)
2026
-
[7]
N. S. H. Gunda and A. Van der Ven, First-principles in- sights on phase stability of titanium interstitial alloys, Phys. Rev. Mater.2, 083602 (2018)
2018
-
[8]
Sarker, T
P . Sarker, T. Harrington, C. Toher, C. Oses, M. Samiee, J.- P . Maria, D. W. Brenner, K. S. Vecchio, and S. Curtarolo, High-entropy high-hardness metal carbides discovered by entropy descriptors, Nature Communications9, 4980 (2018)
2018
-
[9]
D. Dey, L. Liang, and L. Yu, Mixed Enthalpy–Entropy Descriptor for the Rational Design of Synthesizable High- Entropy Materials Over Vast Chemical Spaces, Journal of the American Chemical Society146, 5142 (2024)
2024
-
[10]
Divilov, H
S. Divilov, H. Eckert, D. Hicks, C. Oses, C. Toher, R. Friedrich, M. Esters, M. J. Mehl, A. C. Zettel, Y. Led- erer, E. Zurek, J.-P . Maria, D. W. Brenner, X. Campilongo, S. Filipovi´ c, W. G. Fahrenholtz, C. J. Ryan, C. M. DeSalle, R. J. Crealese, D. E. Wolfe, A. Calzolari, and S. Curtarolo, Disordered enthalpy–entropy descriptor for high-entropy ceramic...
2024
-
[11]
Hedman, A
D. Hedman, A. C. Feltrin, Y. Miyamoto, and F. Akhtar, Ab initio aided design of novel quaternary, quinary and senary high-entropy borocarbides, Journal of Materials Science57, 422 (2022)
2022
-
[12]
M. W. Qureshi, S. Wei, L. Liu, S. Paul, J. Y. Kim, C. Zhang, X. Wang, J. H. Perepezko, D. Morgan, and I. Szlufarska, Predictive screening of phase stability in high-entropy ce- ramics, Materials Advances (2025)
2025
-
[13]
I. Batatia, S. Batzner, D. P . Kovács, A. Musaelian, G. N. C. Simm, R. Drautz, C. Ortner, B. Kozinsky, and G. Csányi, The design space of e(3)-equivariant atom-centered inter- atomic potentials (2022), arXiv:2205.06643
-
[14]
Batatia, D
I. Batatia, D. P . Kovacs, G. N. C. Simm, C. Ortner, and G. Csanyi, MACE: Higher order equivariant message pass- ing neural networks for fast and accurate force fields, in Advances in Neural Information Processing Systems, edited by A. H. Oh, A. Agarwal, D. Belgrave, and K. Cho (2022)
2022
-
[15]
Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Physical Review B99, 014104 (2019)
R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Physical Review B99, 014104 (2019)
2019
-
[16]
D. P . Kovács, I. Batatia, E. S. Arany, and G. Csányi, Evalua- tion of the mace force field architecture: From medicinal chemistry to materials science, The Journal of Chemical Physics159, 044118 (2023)
2023
-
[17]
Kresse and J
G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Physical Review B - Condensed Matter and Materials Physics54, 11169 (1996)
1996
-
[18]
J. P . Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett.77, 3865 (1996)
1996
-
[19]
Kresse, From ultrasoft pseudopotentials to the projector augmented-wave method, Physical Review B - Condensed Matter and Materials Physics59, 1758 (1999)
G. Kresse, From ultrasoft pseudopotentials to the projector augmented-wave method, Physical Review B - Condensed Matter and Materials Physics59, 1758 (1999)
1999
-
[20]
Methfessel and A
M. Methfessel and A. Paxton, High-precision sampling for brillouin-zone integration in metals, Physical Review B40, 3616 (1989)
1989
-
[21]
von Helmholtz,Die Thermodynamik chemischer Vorgänge (Berichte der Königlichen Preussischen Akademie der Wis- senschaften zu Berlin, Berlin, 1882) p
H. von Helmholtz,Die Thermodynamik chemischer Vorgänge (Berichte der Königlichen Preussischen Akademie der Wis- senschaften zu Berlin, Berlin, 1882) p. 22
-
[22]
Y. L. Müller and A. R. Natarajan, First-principles thermo- dynamics of precipitation in aluminum-containing refrac- tory alloys, Acta Materialia274, 119995 (2024)
2024
-
[23]
S. Wei, M. W. Qureshi, J. Wei, L. Liu, X. Hu, J. Xi, S. Attar- ian, R. Su, H. Zhang, E. Willing, X. Wang, K. Sridharan, P . M. Voyles, J. H. Perepezko, and I. Szlufarska, Short- range order in high entropy carbides, Nature Communica- tions17, 2362 (2026)
2026
-
[24]
Zunger, S.-H
A. Zunger, S.-H. Wei, L. G. Ferreira, and J. E. Bernard, Special quasirandom structures, Physical review letters65, 353 (1990)
1990
-
[25]
J. C. Thomas and A. V . d. Ven, Finite-temperature prop- erties of strongly anharmonic and mechanically unstable crystal phases from first principles, Physical Review B88, 214111 (2013)
2013
-
[26]
Puchala and A
B. Puchala and A. Van der Ven, Thermodynamics of the zr-o system from first-principles calculations, Physical Review B—Condensed Matter and Materials Physics88, 094108 (2013)
2013
-
[27]
Van der Ven, J
A. Van der Ven, J. C. Thomas, B. Puchala, and A. R. Natara- jan, First-principles statistical mechanics of multicompo- nent crystals, Annual Review of Materials Research48, 27 (2018)
2018
-
[28]
B. R. Watkins, C. Haas Blacksher, A. Stubbers, G. B. Thomp- son, and C. R. Weinberger, Insights into the anomalous hardness of the tantalum carbides from dislocation mobil- ity, Nature Communications15, 10585 (2024)
2024
-
[29]
B. R. Watkins, Y. Huang, A. Stubbers, G. B. Thompson, and C. R. Weinberger, Plasticity-fracture competition and anomalous hardness in the hard metals, Acta Materialia 298, 121350 (2025)
2025
-
[30]
H. Yu, M. Bahadori, G. B. Thompson, and C. R. Weinberger, Understanding dislocation slip in stoichiometric rocksalt transition metal carbides and nitrides, Journal of Materials Science52, 6235 (2017)
2017
-
[31]
Zhang, L
J. Zhang, L. He, Y. Xiong, S. Huang, B. Xu, S. Ma, X. Xi- ang, H. Fu, J. Kai, Z. Wu, and S. Zhao, Local-distortion- informed exceptional multicomponent transition-metal carbides uncovered by machine learning, npj Computa- tional Materials10, 162 (2024). 13
2024
-
[32]
Mazhnik and A
E. Mazhnik and A. R. Oganov, A model of hardness and fracture toughness of solids, Journal of Applied Physics 126, 125109 (2019)
2019
-
[33]
D. M. Teter, Computational alchemy: The search for new superhard materials, MRS Bulletin23, 22 (1998)
1998
-
[34]
X.-Q. Chen, H. Niu, D. Li, and Y. Li, Modeling hardness of polycrystalline materials and bulk metallic glasses, Inter- metallics19, 1275 (2011)
2011
-
[35]
Y. Tian, B. Xu, and Z. Zhao, Microscopic theory of hard- ness and design of novel superhard crystals, International Journal of Refractory Metals and Hard Materials33, 93 (2012)
2012
- [36]
-
[37]
H. Kaur, F. Della Pia, I. Batatia, X. R. Advincula, B. X. Shi, J. Lan, G. Csányi, A. Michaelides, and V . Kapil, Data-efficient fine-tuning of foundational models for first- principles quality sublimation enthalpies, Faraday Discuss. 256, 120 (2025)
2025
-
[38]
Radova, W
M. Radova, W. G. Stark, C. S. Allen, R. J. Maurer, and A. P . Bartók, Fine-tuning foundation models of materials interatomic potentials with frozen transfer learning, npj Computational Materials11, 237 (2025)
2025
-
[39]
L. Piersante and A. R. Natarajan, Machine learning in- teratomic potentials for solid-state precipitation (2026), arXiv:2601.12984 [cond-mat.mtrl-sci]
-
[40]
A. R. Natarajan, P . Dolin, and A. Van der Ven, Crystallogra- phy, thermodynamics and phase transitions in refractory binary alloys, Acta Materialia200, 171 (2020)
2020
-
[41]
M. D. Hossain, T. Borman, C. Oses, M. Esters, C. To- her, L. Feng, A. Kumar, W. G. Fahrenholtz, S. Curtarolo, D. Brenner,et al., Entropy landscaping of high-entropy carbides, Advanced Materials33, 2102904 (2021)
2021
-
[42]
Castle, T
E. Castle, T. Csanádi, S. Grasso, J. Dusza, and M. Reece, Processing and properties of high-entropy ultra-high tem- perature carbides, Scientific reports8, 8609 (2018)
2018
-
[43]
T. J. Harrington, J. Gild, P . Sarker, C. Toher, C. M. Rost, O. F. Dippo, C. McElfresh, K. Kaufmann, E. Marin, L. Borowski, P . E. Hopkins, J. Luo, S. Curtarolo, D. W. Brenner, and K. S. Vecchio, Phase stability and mechanical properties of novel high entropy transition metal carbides, Acta Materialia166, 271 (2019)
2019
-
[44]
Chicardi, C
E. Chicardi, C. García-Garrido, and F. Gotor, Low tempera- ture synthesis of an equiatomic (tizrhfvnb) c5 high entropy carbide by a mechanically-induced carbon diffusion route, Ceramics International45, 21858 (2019)
2019
-
[45]
Wei, J.-X
X.-F. Wei, J.-X. Liu, F. Li, Y. Qin, Y.-C. Liang, and G.-J. Zhang, High entropy carbide ceramics from different start- ing materials, Journal of the European Ceramic Society39, 2989 (2019)
2019
-
[46]
Chicardi, C
E. Chicardi, C. García-Garrido, J. Hernández-Saz, and F. Gotor, Synthesis of all equiatomic five-transition metals high entropy carbides of the ivb (ti, zr, hf) and vb (v, nb, ta) groups by a low temperature route, Ceramics International 46, 21421 (2020)
2020
-
[47]
Raju Natarajan and A
A. Raju Natarajan and A. Van der Ven, Toward an Under- standing of Deformation Mechanisms in Metallic Lithium and Sodium from First-Principles, Chemistry of Materials 31, 8222 (2019)
2019
-
[48]
Baruffi, M
C. Baruffi, M. Ghazisaeidi, D. Rodney, and W. Curtin, Equi- librium versus non-equilibrium stacking fault widths in nicocr, Scripta Materialia235, 115536 (2023)
2023
-
[49]
P . Li, C. Zhao, Y. Jiang, F. Cao, P . Xiao, Y. Song, Z. Hong, S. Gou, and S. Liang, The relationship between deforma- tion mechanisms and mechanical properties in nanocrys- talline cu/ag-bilayer alloy, Journal of Alloys and Com- pounds986, 174091 (2024)
2024
-
[50]
Kaufmann, D
K. Kaufmann, D. Maryanovsky, W. M. Mellor, C. Zhu, A. S. Rosengarten, T. J. Harrington, C. Oses, C. Toher, S. Cur- tarolo, and K. S. Vecchio, Discovery of high-entropy ce- ramics via machine learning, npj Computational Materials 6, 42 (2020)
2020
-
[51]
Varvenne, A
C. Varvenne, A. Luque, and W. A. Curtin, Theory of strengthening in fcc high entropy alloys, Acta Materialia 118, 164 (2016)
2016
-
[52]
Varvenne, G
C. Varvenne, G. Leyson, M. Ghazisaeidi, and W. Curtin, Solute strengthening in random alloys, Acta Materialia 124, 660 (2017)
2017
-
[53]
Liu and W
X. Liu and W. Curtin, Atomistic simulations reveal strength reductions due to short-range order in alloys, Acta Materialia263, 119471 (2024)
2024
-
[54]
high- entropy
F. Maresca and W. A. Curtin, Theory of screw dislocation strengthening in random bcc alloys from dilute to “high- entropy” alloys, Acta Materialia182, 144 (2020)
2020
-
[55]
Maresca and W
F. Maresca and W. A. Curtin, Mechanistic origin of high strength in refractory bcc high entropy alloys up to 1900k, Acta Materialia182, 235 (2020)
2020
-
[56]
Synthesizability, hardness, and stacking order in multicomponent transition metal carbides from machine-learned potentials
S. Karumuri, A. Hernandez, S. Mishra, Z. McClure, V . Tucker, J. C. Flanagan, S. Hwang, K. H. Sandhage, I. Bil- ionis, M. S. Titus, and A. Strachan, Design of high-hardness complex concentrated alloys from physics, machine learn- ing, and experiments, Journal of Applied Physics138, 085106 (2025). Supplementary Materials for “Synthesizability, hardness, an...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.