Adaptive fine-tuning of foundation models for crystal structure prediction: Discovery of high-pressure phases in the CaFeNi system
Pith reviewed 2026-07-01 01:23 UTC · model grok-4.3
The pith
Adaptive fine-tuning workflow predicts new Ca6FeNi compound stable above 100 GPa in Ca-Fe-Ni system.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
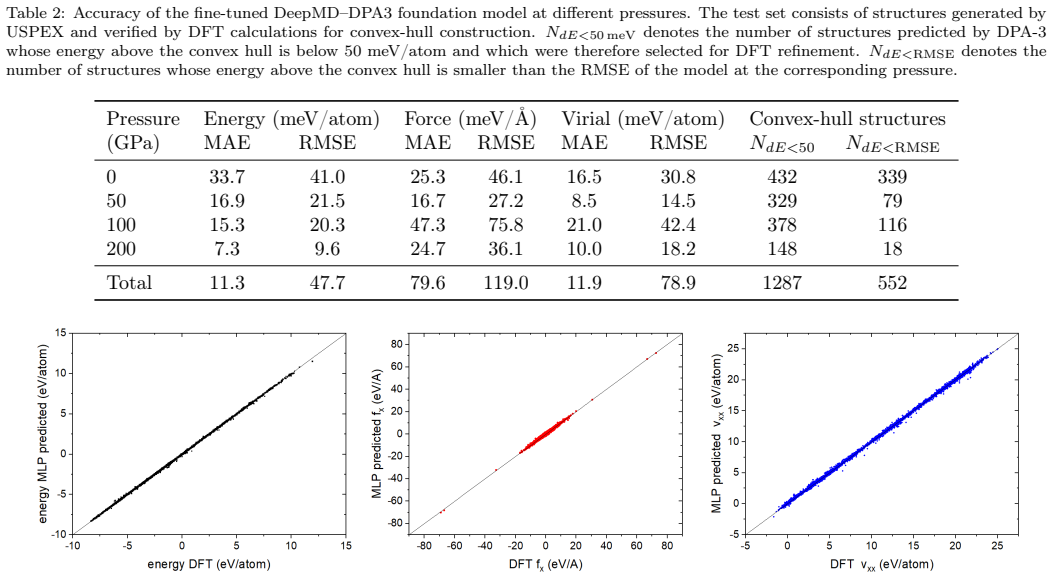
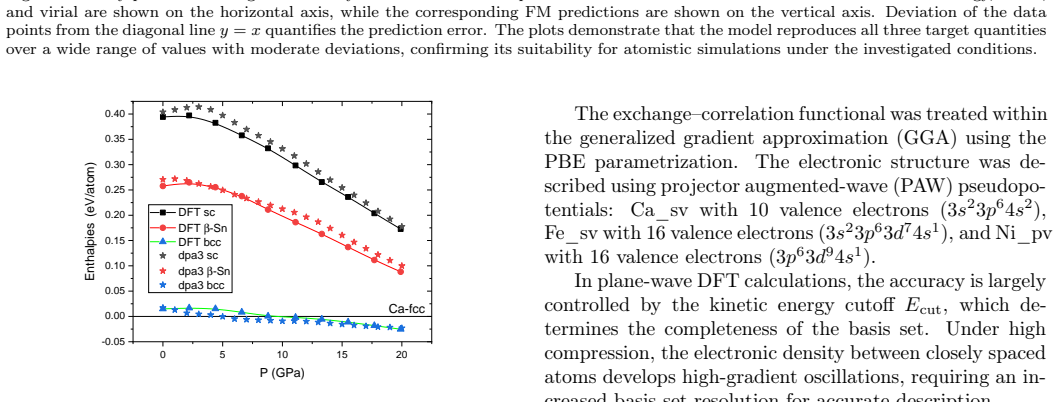
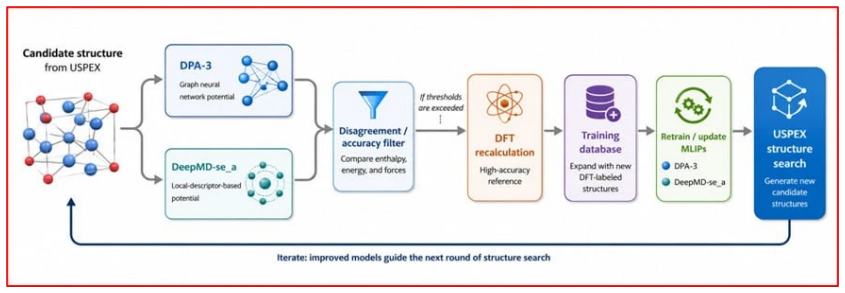
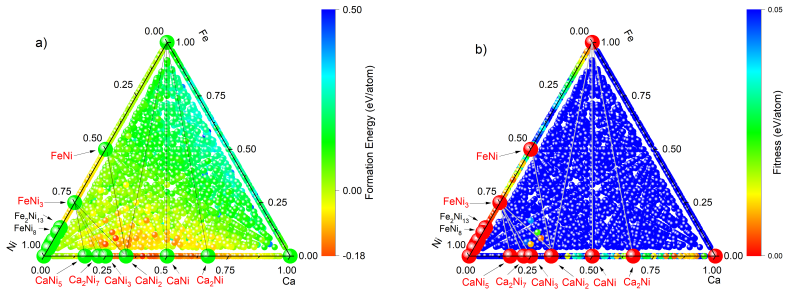
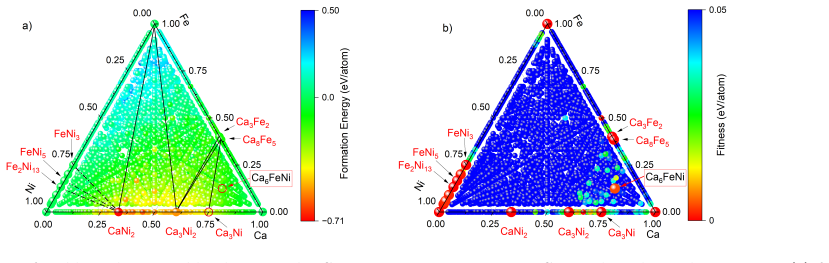
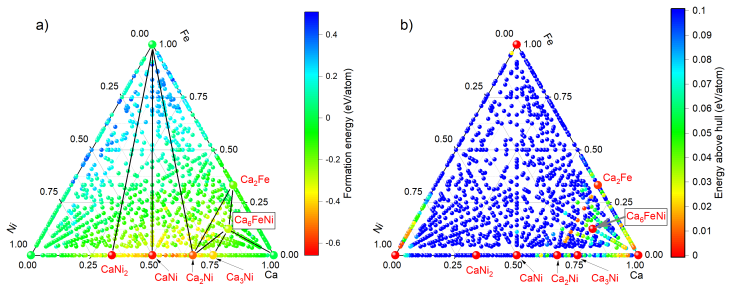
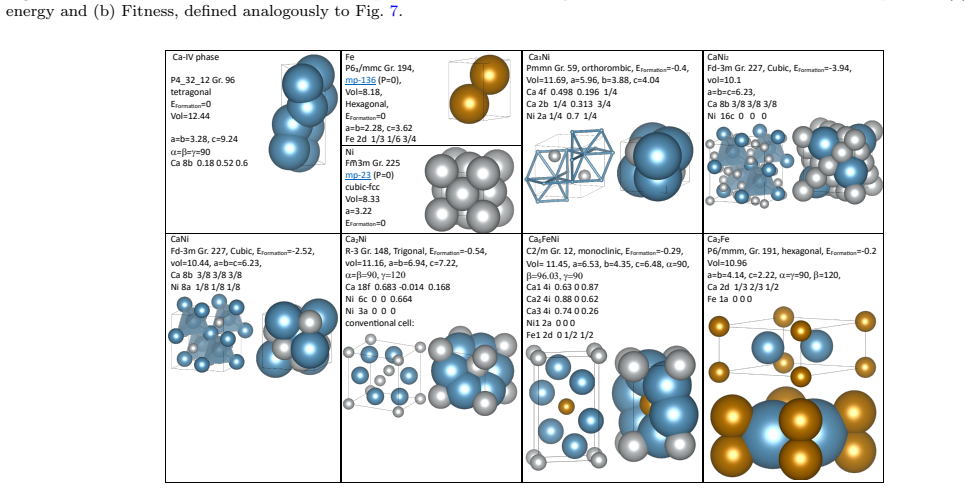
The workflow reproduces the known low-pressure convex hull of the Ca-Fe-Ni system and predicts that the unreported compound Ca6FeNi becomes thermodynamically stable above 100 GPa. It achieves this by combining evolutionary search with adaptive data selection that chooses compact, representative subsets of structures for DFT labeling, then fine-tunes the foundation-model potential on those labels to improve energy predictions for the target chemical space.
What carries the argument
Adaptive data selection for iterative fine-tuning of a pretrained MLIP, which identifies compact representative structure subsets for DFT labeling to build a system-specific potential that ranks energies for stability assessment.
If this is right
- The workflow reproduces the known low-pressure convex hull for the Ca-Fe-Ni system.
- It enables efficient exploration of high-pressure phases without large initial training sets.
- It identifies Ca6FeNi as a new compound stable above 100 GPa.
- Computational cost drops while accuracy for energy rankings is preserved in multicomponent systems.
- The method extends to other chemically complex ternary and higher systems.
Where Pith is reading between the lines
- The same selection and fine-tuning loop could be tested on other ternary systems to locate additional pressure-induced phases.
- Efficiency gains might allow the workflow to incorporate experimental constraints or couple to molecular dynamics for kinetic stability checks.
- The approach implies that general foundation models can be specialized for specific chemical spaces with far fewer labels than training from scratch.
Load-bearing premise
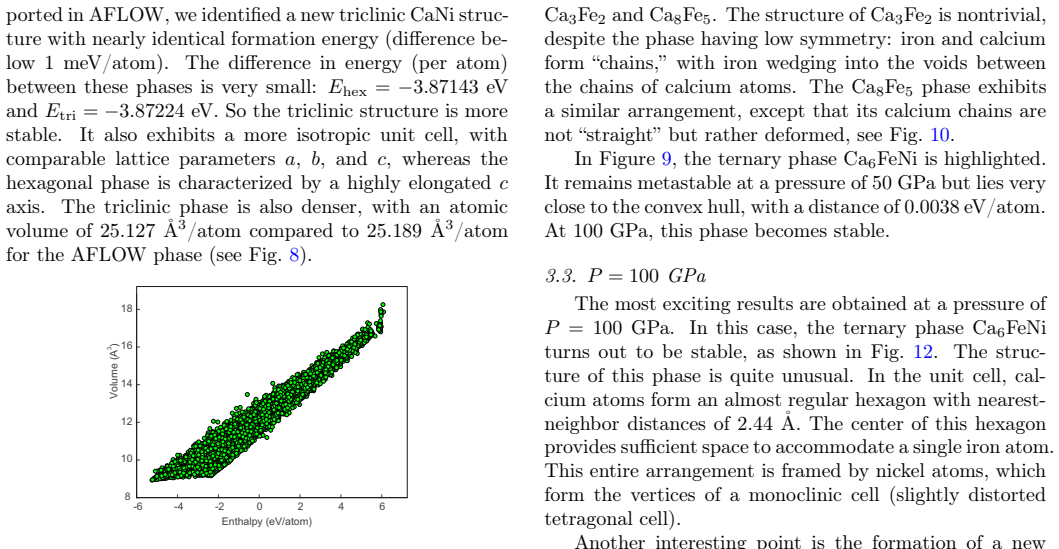
The adaptive selection produces subsets whose DFT labels allow the fine-tuned potential to correctly rank energies and determine thermodynamic stability for high-pressure configurations in this ternary system.
What would settle it
Full DFT relaxation and energy calculation of the predicted Ca6FeNi structure placed on the convex hull at pressures above 100 GPa, confirming or refuting its stability relative to competing phases.
Figures
read the original abstract
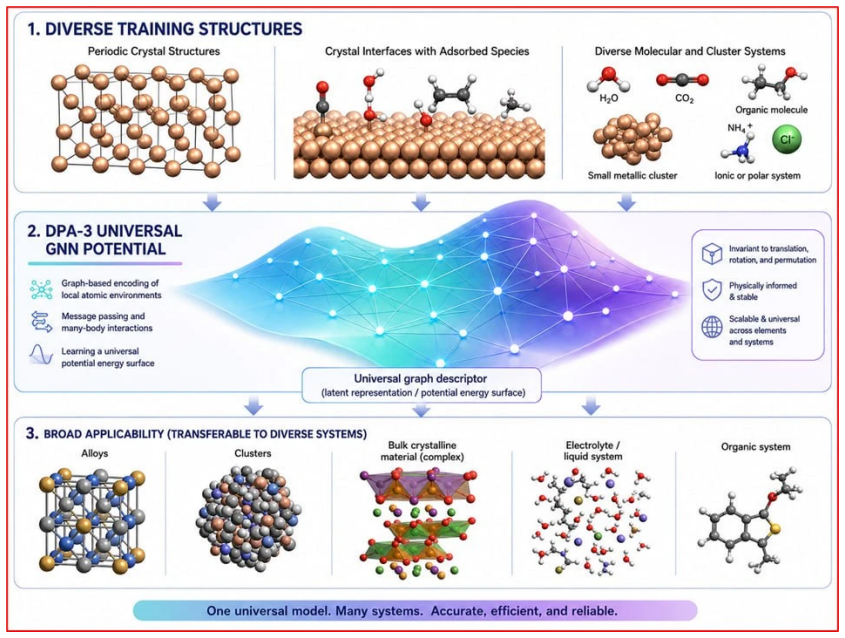
The prediction of crystal structures is a key challenge in chemistry and materials science, but evolutionary crystal structure prediction (CSP) remains computationally expensive because it relies on repeated \textit{ab initio} relaxations and energy ranking. Machine learning interatomic potentials (MLIPs) can accelerate CSP, yet their use is limited by the need for large training sets and by the difficulty of choosing which candidate structures should be labeled by density functional theory (DFT). Here we introduce a self-consistent, foundation-model-assisted CSP workflow that combines evolutionary search with adaptive data selection and fine-tuning. Starting from a pretrained MLIP, the algorithm rapidly explores configuration space while iteratively selecting compact, representative, and physically relevant subsets of structures for DFT labeling, thereby reducing redundant calculations and improving a system-specific potential. We apply the method to the chemically complex Ca--Fe--Ni ternary system. The workflow reproduces the known low-pressure convex hull and enables efficient high-pressure exploration. It predicts a previously unreported compound, Ca$_6$FeNi, which becomes thermodynamically stable above 100~GPa. These results show that foundation-model-based, data-efficient CSP can greatly reduce computational cost while preserving accuracy and enabling the discovery of new materials in complex multicomponent systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a self-consistent CSP workflow that starts from a pretrained foundation-model MLIP, uses evolutionary search, and iteratively applies adaptive data selection to choose compact subsets of structures for DFT labeling and fine-tuning. Applied to the Ca-Fe-Ni ternary, the workflow reproduces the known low-pressure convex hull and identifies a previously unreported compound, Ca6FeNi, that is predicted to lie on the convex hull above 100 GPa.
Significance. If the central validation claim holds, the adaptive fine-tuning approach would represent a meaningful advance in data-efficient CSP for chemically complex systems, reducing the number of DFT evaluations while enabling high-pressure exploration. The paper explicitly credits the foundation-model starting point and the iterative selection for compactness, which are genuine strengths if the energy rankings are shown to be reliable for the reported discovery.
major comments (2)
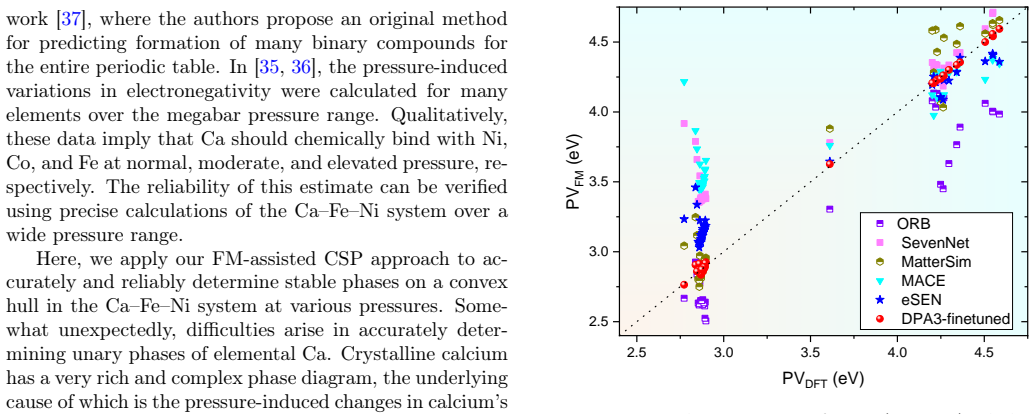
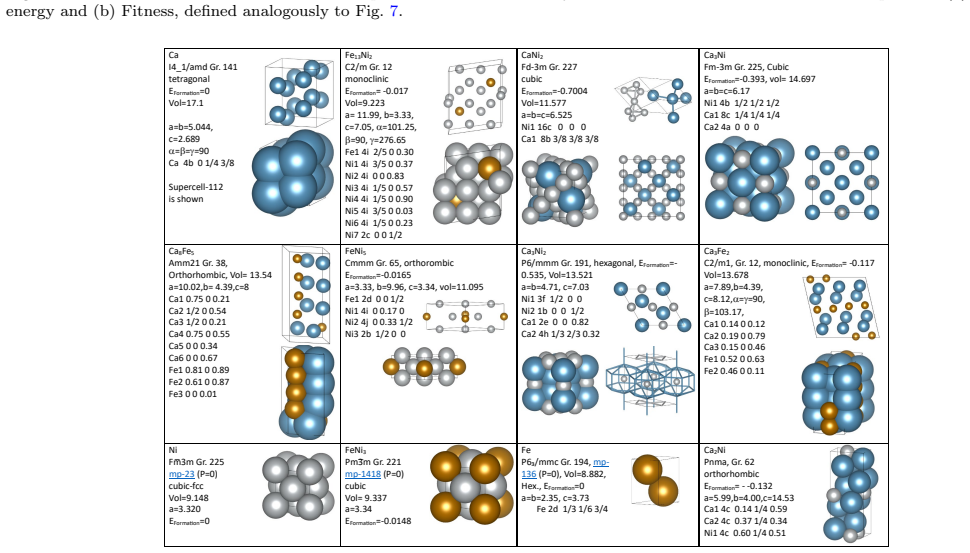
- [§4] §4 (high-pressure results): the placement of Ca6FeNi on the convex hull above 100 GPa is determined exclusively from the fine-tuned MLIP energies; no table or figure reports direct DFT formation energies or hull distances for the final candidate structures at those pressures, leaving the key stability claim without an independent check.
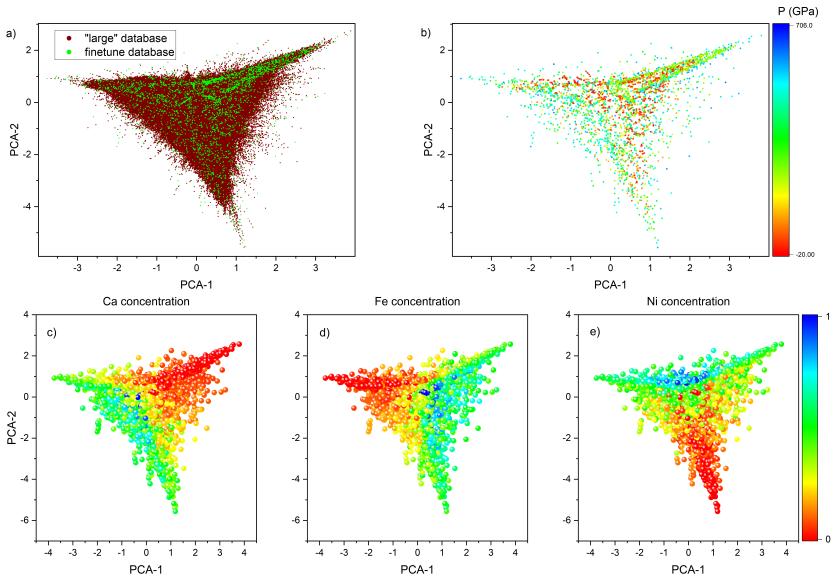
- [§3.2] §3.2 (adaptive selection): the selection metric is described as producing representative subsets, but no quantitative breakdown (e.g., fraction of high-P ternary vs. binary or low-P structures) or uncertainty quantification on the selected high-pressure configurations is given, so it is unclear whether the training set bounds the extrapolation error for the reported ternary hull.
minor comments (2)
- [Figure 3] Figure 3 caption should explicitly state the pressure range and number of DFT labels used for the high-P hull construction.
- [Abstract] The abstract states that the workflow 'reproduces the known low-pressure convex hull' but does not cite the specific known phases or provide a quantitative match metric (e.g., formation-energy MAE).
Simulated Author's Rebuttal
We thank the referee for the constructive review. We address each major comment below and will revise the manuscript accordingly to strengthen the validation and clarity of the adaptive workflow.
read point-by-point responses
-
Referee: [§4] §4 (high-pressure results): the placement of Ca6FeNi on the convex hull above 100 GPa is determined exclusively from the fine-tuned MLIP energies; no table or figure reports direct DFT formation energies or hull distances for the final candidate structures at those pressures, leaving the key stability claim without an independent check.
Authors: We acknowledge that the final high-pressure hull placement for Ca6FeNi relies on the fine-tuned MLIP. Although DFT labeling occurs throughout the adaptive iterations, we agree an independent check strengthens the claim. In the revised manuscript we will add direct DFT single-point calculations (and hull distances) for the key Ca6FeNi and competing structures at 100 GPa and 150 GPa, together with a comparison table of MLIP versus DFT formation energies. This will provide the requested validation while preserving the data-efficient nature of the workflow. revision: yes
-
Referee: [§3.2] §3.2 (adaptive selection): the selection metric is described as producing representative subsets, but no quantitative breakdown (e.g., fraction of high-P ternary vs. binary or low-P structures) or uncertainty quantification on the selected high-pressure configurations is given, so it is unclear whether the training set bounds the extrapolation error for the reported ternary hull.
Authors: We agree that quantitative details on the adaptive selection would improve transparency. The revised manuscript will include a table (or supplementary figure) breaking down the selected structures by pressure regime and composition type (e.g., percentage of high-P ternary versus binary or low-P structures). We will also report ensemble-based uncertainty estimates on the high-pressure configurations to demonstrate that extrapolation error is bounded for the ternary hull. revision: yes
Circularity Check
No significant circularity; workflow outputs are independent of inputs
full rationale
The paper presents an iterative MLIP fine-tuning workflow for CSP that selects structures for DFT labeling and uses the resulting potential to rank energies and identify hull membership for Ca6FeNi above 100 GPa. No quoted step equates a prediction to a fitted input by construction, renames a known result, or reduces the central stability claim to a self-citation chain or self-definition. The adaptive selection and energy ranking are presented as producing new information about high-pressure configurations, with the discovery framed as an empirical output rather than a tautology. The derivation chain remains self-contained against external DFT benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
A. R. Oganov, C. J. Pickard, Q. Zhu, R. J. Needs, Structure prediction drives materials discovery, Nature Reviews Materials 4 (2019) 331–348.doi:10.1038/s41578-019-0101-8
-
[2]
S. M. Woodley, R. Catlow, Crystal structure prediction from first principles, Nature Materials 7 (2008) 937–946.doi:10. 1038/nmat2321
2008
-
[3]
Zurek, W
E. Zurek, W. Grochala, Predicting crystal structures and prop- erties of matter under extreme conditions, Physical Chem- istry Chemical Physics 17 (2015) 2917–2934.doi:10.1039/ C4CP04445B
2015
-
[4]
Y. Wang, Y. Ma, Perspective: Crystal structure prediction at high pressures, Journal of Chemical Physics 140 (2014) 040901. doi:10.1063/1.4861966
-
[5]
A. R. Oganov, C. W. Glass, Crystal structure prediction using ab initio evolutionary techniques, Journal of Chemical Physics 124 (2006) 244704.doi:10.1063/1.2210932
-
[6]
Y. Wang, J. Lv, Q. Zhu, Y. Ma, Calypso: A method for crystal structure prediction, Computer Physics Communications 183 (2012) 2063–2070.doi:10.1016/j.cpc.2012.05.008
-
[7]
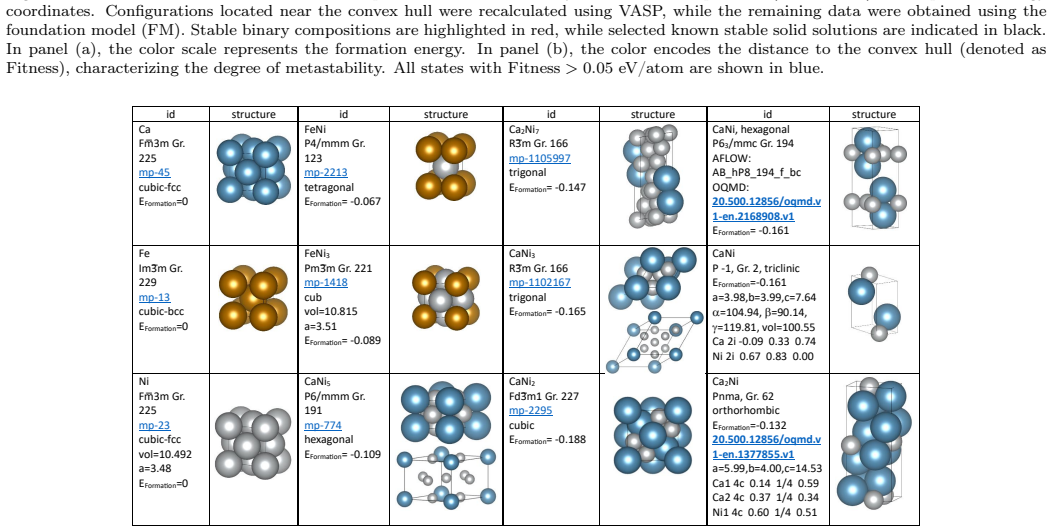
A. O. Lyakhov, A. R. Oganov, H. T. Stokes, Q. Zhu, New de- velopments in evolutionary structure prediction algorithm us- pex, Computer Physics Communications 184 (2013) 1172–1182. doi:10.1016/j.cpc.2012.12.009. 11 Figure 12: Distribution of stable and metastable phases in the Ca–Fe–Ni system atP= 100GPa. The color scale represents (a) formation energy and...
-
[8]
I. A. Kruglov, A. V. Yanilkin, Y. Propad, A. B. Mazitov, P. Rachitskii, A. R. Oganov, Crystal structure prediction at finite temperatures, npj Computational Materials 9 (2023) 197. doi:10.1038/s41524-023-01120-6
-
[9]
J. Behler, Perspective: Machine learning potentials for atom- istic simulations, Journal of Chemical Physics 145 (2016) 170901.doi:10.1063/1.4966192
-
[10]
O. T. Unke, et al., Machine learning force fields, Chemical Reviews 121 (2021) 10142–10186.doi:10.1021/acs.chemrev. 0c01111
-
[11]
L. Zhang, et al., Active learning of uniformly accurate inter- atomic potentials, Physical Review Materials 3 (2019) 023804. doi:10.1103/PhysRevMaterials.3.023804
-
[12]
J. S. Smith, et al., Less is more: Sampling chemical space with active learning, Journal of Chemical Physics 148 (2018) 241733. doi:10.1063/1.5023802
-
[13]
M. Kellner, M. Ceriotti, Uncertainty quantification by direct propagation of shallow ensembles, Machine Learning: Science and Technology 5 (3) (2024) 035006.doi:10.1088/2632-2153/ ad594a
-
[14]
F. Bigi, S. Chong, M. Ceriotti, F. Grasselli, A prediction rigidity formalism for low-cost uncertainties in trained neural networks, Machine Learning: Science and Technology 5 (4) (2024) 045018. doi:10.1088/2632-2153/ad805f
-
[15]
I. Batatia, et al., Mace: Higher-order equivariant message pass- ing neural networks, Advances in Neural Information Processing Systems (2022).doi:10.48550/arXiv.2206.07697
-
[16]
Batatia, et al., A general framework to learn invari- ant and equivariant representations of atomistic systems, Nature Communications 14 (2023) 2453.doi:10.1038/ s41467-023-38192-7
I. Batatia, et al., A general framework to learn invari- ant and equivariant representations of atomistic systems, Nature Communications 14 (2023) 2453.doi:10.1038/ s41467-023-38192-7
2023
-
[18]
I. Batatia, et al., The mace-off: Benchmarking foundation mod- els for interatomic potentials, arXiv (2024).doi:10.48550/ arXiv.2405.12345
-
[19]
A. M. Dziewonski, D. L. Anderson, Preliminary reference earth model, Physics of the Earth and Planetary Interi- ors 25 (4) (1981) 297–356.doi:https://doi.org/10.1016/ 0031-9201(81)90046-7. URLhttps://www.sciencedirect.com/science/article/pii/ 0031920181900467
1981
-
[20]
W. McDonough, S. s. Sun, The composition of the earth, Chemical Geology 120 (3) (1995) 223–253, chemical Evolution of the Mantle.doi:https://doi.org/10.1016/0009-2541(94) 12 00140-4. URLhttps://www.sciencedirect.com/science/article/pii/ 0009254194001404
-
[21]
K. Litasov, A. Shatskiy, Composition of the earth’s core: A review, Russian Geology and Geophysics 57 (1) (2016) 22–46.arXiv:https://pubs.geoscienceworld.org/nsu/rgg/ article-pdf/57/1/22/5919580/v57i1_s1068797116000043. pdf,doi:10.1016/j.rgg.2016.01.003. URLhttps://doi.org/10.1016/j.rgg.2016.01.003
-
[22]
B. J. Wood, M. J. Walter, J. Wade, Accretion of the earth and segregation of its core, Nature 441 (2006) 825–833.doi:https: //doi.org/10.1038/nature04763. URLhttps://doi.org/10.1038/nature04763
-
[23]
K. Watanabe, E. Ohtani, S. Kamada, T. Sakamaki, M. Miya- hara, Y. Ito, The abundance of potassium in the earth’s core, Physics of the Earth and Planetary Interiors 237 (2014) 65–72. doi:https://doi.org/10.1016/j.pepi.2014.10.001. URLhttps://www.sciencedirect.com/science/article/pii/ S003192011400212X
-
[24]
L. J. Parker, T. Atou, J. V. Badding, Transition element-like chemistry for potassium under pressure, Science 273 (5271) (1996) 95–97.doi:https://www.science.org/doi/10.1126/ science.273.5271.95
1996
-
[25]
A. V. Tsvyashchenko, L. N. Fomicheva, M. V. Magnitskaya, V. A. Sidorov, A. V. Kuznetsov, D. V. Eremenko, V. N. Trofimov, New ferromagnetic compound caco2 (c15) synthe- sized at high pressure, Journal of Experimental and Theo- retical Physics Letters 68 (12) (1998) 864–869.doi:https: //doi.org/10.1134/1.567954. URLhttp://jetpletters.ru/ps/972/article_14815.pdf
-
[26]
A. V. Tsvyashchenko, L. N. Fomicheva, M. V. Magnitskaya, V. A. Sidorov, E. N. Shirani, A. V. Kuznetsov, D. V. Ere- menko, V. H. Trofimov, Magnetism of ca–3d metal laves-phase compounds synthesized at high pressure, Physics of Metals and Metallography 93 (2002) S59 – S63
2002
-
[27]
H. Yan, J. Shi, S. Hou, From deep earth to deep stars: A differ- ent nuclear reaction path to generate calcium in ancient stars, The Innovation 4 (1) (2023) 100377.doi:https://doi.org/10. 1016/j.xinn.2023.100377. URLhttps://www.sciencedirect.com/science/article/pii/ S266667582300005X
-
[28]
G. X. Dong, X. B. Wang, N. Michel, M. P loszajczak, 19F(p, γ)20Nereaction rate and the puzzling calcium abun- dance in metal-poor stars, Phys. Rev. C 110 (6) (2024) L061601.doi:10.1103/PhysRevC.110.L061601. URLhttps://link.aps.org/doi/10.1103/PhysRevC.110. L061601
-
[29]
E. G. Maksimov, M. V. Magnitskaya, V. E. Fortov, Non-simple behavior of simple metals at high pressure, Physics-Uspekhi 48 (8) (2005) 761–780.doi:10.1070/ pu2005v048n08abeh002315. URLhttps://ufn.ru/en/articles/2005/8/a/
2005
-
[30]
A. R. Miedema, On the heat of formation of solid alloys, Journal of the Less Common Metals 41 (2) (1975) 283–298.doi:https: //doi.org/10.1016/0022-5088(75)90034-X. URLhttps://www.sciencedirect.com/science/article/abs/ pii/002250887590034X
-
[31]
Pettifor, Bonding and Structure of Molecules and Solids, Clarendon Press, 1995
D. Pettifor, Bonding and Structure of Molecules and Solids, Clarendon Press, 1995. URLhttps://books.google.ru/books?id=r7XGPHD24fgC
1995
-
[32]
M. Rahm, R. Cammi, N. W. Ashcroft, R. Hoffmann, Squeezing all elements in the periodic table: Electron configuration and electronegativity of the atoms under compression, Journal of the American Chemical Society 141 (26) (2019) 10253–10271. doi:10.1021/jacs.9b02634. URLhttps://doi.org/10.1021/jacs.9b02634
-
[33]
M. Rahm, T. Zeng, R. Hoffmann, Electronegativity seen as the ground-state average valence electron binding energy, Journal of the American Chemical Society 141 (1) (2019) 342–351.doi: 10.1021/jacs.8b10246. URLhttps://doi.org/10.1021/jacs.8b10246
-
[34]
Z. Allahyari, A. R. Oganov, Nonempirical definition of the mendeleev numbers: Organizing the chemical space, The Jour- nal of Physical Chemistry C 124 (43) (2020) 23867–23878. doi:https://doi.org/10.1021/acs.jpcc.0c07857
-
[35]
M. Rahm, P. Erhart, R. Cammi, Relating atomic energy, radius and electronegativity through compression, Chemical Science 12 (7) (2021) 2397–2403.doi:10.1039/D0SC06675C. URLhttp://dx.doi.org/10.1039/D0SC06675C
-
[36]
X. Dong, A. R. Oganov, H. Cui, X.-F. Zhou, H.-T. Wang, Electronegativity and chemical hardness of elements under pressure, Proceedings of the National Academy of Sciences 119 (10) (2022) e2117416119.doi:https://doi.org/10.1073/ pnas.2117416119
2022
-
[37]
A. R. Oganov, M. G. Kostenko, Simple electronegativity-based model for predicting formation of stable compounds across the periodic table, Nature Communications 17 (1) (2025) 929.doi: https://doi.org/10.1038/s41467-025-67658-9
-
[38]
X. Dong, A. R. Oganov, Electrides and their high-pressure chemistry, in: G. G. N. Angilella, A. La Magna (Eds.), Cor- relations in Condensed Matter under Extreme Conditions: A tribute to Renato Pucci on the occasion of his 70th birth- day, Springer International Publishing, Cham, 2017, pp. 69–84. doi:10.1007/978-3-319-53664-4_6. URLhttps://doi.org/10.1007...
-
[39]
D. Y. Novoselov, D. M. Korotin, A. O. Shorikov, A. R. Oganov, V. I. Anisimov, Weak coulomb correlations stabilize the electride high-pressure phase of elemental calcium, Jour- nal of Physics: Condensed Matter 32 (44) (2020) 445501. doi:10.1088/1361-648X/ab99ed. URLhttps://doi.org/10.1088/1361-648X/ab99ed
-
[40]
P. Modak, A. K. Verma, P. M. Oppeneer, Pressure-induced electride phase formation in calcium: A key to its strange high-pressure behavior, Phys. Rev. B 107 (12) (2023) 125152. doi:10.1103/PhysRevB.107.125152. URLhttps://link.aps.org/doi/10.1103/PhysRevB.107. 125152
-
[41]
S. Racioppi, E. Zurek, Looking at high-pressure electrides through the lens of quantum crystallography: the case of simple cubic calcium, Acta Crystallographica Section B 81 (2) (2025) 256–265.doi:10.1107/S2052520625001647. URLhttps://doi.org/10.1107/S2052520625001647
-
[42]
M. Hirayama, R. Okugawa, T. Miyake, S. Murakami, Topo- logical dirac nodal lines and surface charges in fcc alkaline earth metals, Nature Communications 8 (1) (2017) 14022.doi: 10.1038/ncomms14022. URLhttps://doi.org/10.1038/ncomms14022
-
[43]
V. V. Brazhkin, O. B. Tsiok, M. V. Magnitskaya, Thermoelec- tric power of calcium at high pressure, JETP Letters 83 (7) (2006) 377–381.doi:10.1134/S0021364006070029. URLhttps://jetpletters.ru/ps/2006/article_30278.pdf
-
[44]
M. V. Magnitskaya, N. L. Matsko, V. S. Baturin, Y. A. Uspen- skii, Pressure-induced semimetallic behavior of calcium from ab initio calculations, Journal of Physics: Conference Series 510 (1) (2014) 012028.doi:10.1088/1742-6596/510/1/012028. URLhttps://dx.doi.org/10.1088/1742-6596/510/1/012028
-
[45]
A. R. Oganov, Y. Ma, Y. Xu, I. Errea, A. Bergara, A. O. Lyakhov, Exotic behavior and crystal structures of calcium un- der pressure, Proceedings of the National Academy of Sciences 107 (17) (2010) 7646–7651.doi:10.1073/pnas.0910335107. URLhttps://www.pnas.org/doi/abs/10.1073/pnas. 0910335107
-
[46]
S. Arapan, H. kwang Mao, R. Ahuja, Prediction of incommen- surate crystal structure in ca at high pressure, Proceedings of the National Academy of Sciences 105 (52) (2008) 20627–20630. doi:10.1073/pnas.0810813105. URLhttps://www.pnas.org/doi/abs/10.1073/pnas. 0810813105
-
[47]
A. Jain, S. P. Ong, G. Hautier, et al., Commentary: The materials project: A materials genome approach to acceler- ating materials innovation, APL Materials 1 (2013) 011002. doi:10.1063/1.4812323. 13
-
[48]
A. Merchant, et al., Alexandria: A foundation model for atom- istic materials simulations, arXiv (2024).doi:10.48550/arXiv. 2401.00000
work page internal anchor Pith review doi:10.48550/arxiv 2024
-
[49]
O. M. Team, Omat24: A large-scale dataset for universal in- teratomic potentials, arXiv (2024).doi:10.48550/arXiv.2402. 00000
-
[50]
J. Riebesell, R. E. A. Goodall, P. Benner, Y. Chiang, B. Deng, G. Ceder, M. Asta, A. A. Lee, A. Jain, K. A. Persson, A framework to evaluate machine learning crystal stability pre- dictions, Nature Machine Intelligence 7 (6) (2025) 836–847. doi:10.1038/s42256-025-01055-1
-
[51]
L. Zhang, J. Han, H. Wang, W. A. Saidi, R. Car, W. E, Deep potential molecular dynamics: A scalable model with the accu- racy of quantum mechanics, Physical Review Letters 120 (2018) 143001.doi:10.1103/PhysRevLett.120.143001
-
[52]
H. Wang, L. Zhang, J. Han, W. E, Deepmd-kit: A deep learning package for many-body potential energy representa- tion, Computer Physics Communications 228 (2020) 178–184. doi:10.1016/j.cpc.2018.03.016
-
[53]
URLhttps://matbench-discovery.materialsproject.org
Matbench discovery, accessed: 2025-02-13 (2025). URLhttps://matbench-discovery.materialsproject.org
2025
-
[54]
B. Cheng, R.-R. Griffiths, S. Wengert, C. Kunkel, T. Stenczel, B. Zhu, V. L. Deringer, N. Bernstein, J. T. Margraf, K. Reuter, G. Csanyi, Mapping materials and molecules, Accounts of Chemical Research 53 (9) (2020) 1981–1991.doi:10.1021/acs. accounts.0c00403. URLhttps://doi.org/10.1021/acs.accounts.0c00403
work page doi:10.1021/acs 2020
- [55]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.