When Molecular Similarity Works: Property Cliffs Reveal Hidden Errors
Pith reviewed 2026-05-20 14:11 UTC · model grok-4.3
The pith
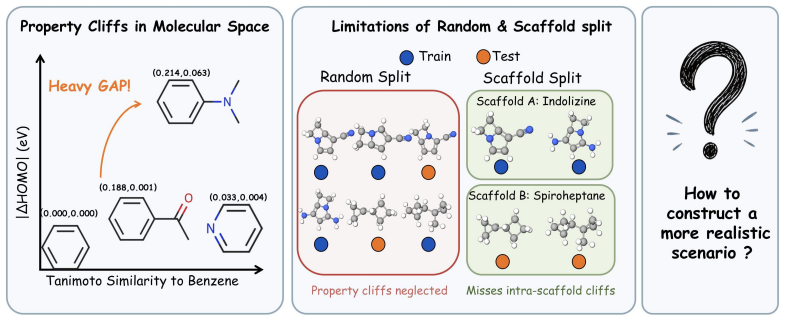
Property cliffs expose hidden errors in molecular property prediction that overall metrics miss.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Property cliffs expose a gap in standard evaluation for molecular machine learning: models with competitive overall performance fail in high-risk local neighborhoods where similar molecules differ sharply in target property. CliffSplit constructs locally supported, cliff-exposed test cases to quantify this, revealing at least 15% higher error in cliff-heavy QM9 regions. CliffLoss, a train-only mechanism, reduces the cliff-to-smooth error gap by up to 30% on Lipophilicity and improves overall MAE by 9.7%.
What carries the argument
CliffSplit, a cliff-aware evaluation protocol that constructs locally supported, cliff-exposed test cases, together with CliffLoss, a model-agnostic train-only mitigation mechanism for cliff-sensitive errors.
If this is right
- Overall performance numbers can conceal large local errors in neighborhoods where molecular similarity breaks down.
- CliffSplit testing uncovers at least 15% higher error in cliff-heavy portions of QM9 targets.
- CliffLoss training shrinks the cliff-to-smooth error difference by as much as 30% on tasks like Lipophilicity.
- The combination turns an anecdotal observation about similarity failure into a measurable benchmark for molecular models.
Where Pith is reading between the lines
- The same cliff-aware split and loss ideas could be tested on other structure-to-property tasks outside small molecules.
- Active learning pipelines might use cliff detection to select additional data points from high-risk neighborhoods.
- Combining CliffLoss with uncertainty estimates could further improve reliability in safety-critical molecular design.
Load-bearing premise
The chosen similarity metric and neighborhood definition correctly identify regions where molecular similarity should predict property similarity but does not.
What would settle it
If models evaluated on the cliff-exposed test sets from CliffSplit show no measurable error increase relative to standard random splits, the claim of undetected localized failures would be refuted.
Figures









read the original abstract
Accurate prediction of molecular properties underpins drug discovery and material design, yet even state-of-the-art models remain vulnerable to localized failure modes that aggregate metrics cannot detect. The places where molecular similarity should be most helpful are also places where standard evaluation can be most misleading. Property cliffs expose this gap: structurally similar molecules can still differ sharply in target property, so models with competitive overall performance may fail in high-risk local neighborhoods. To expose and mitigate this failure mode, CliffSplit, a cliff-aware evaluation protocol that constructs locally supported, cliff-exposed test cases, and CliffLoss, a model-agnostic train-only mitigation mechanism for cliff-sensitive errors, are introduced. Experiments on three QM9 targets and three MoleculeNet tasks across five backbones show that CliffSplit reveals at least 15% higher error in cliff-heavy QM9 regions, while CliffLoss reduces the cliff-to-smooth error gap by up to 30% on Lipophilicity and improves overall MAE by 9.7%. Together, these results turn molecular similarity failure from a descriptive anomaly into a benchmarked evaluation problem for molecular machine learning. The code is available at https://anonymous.4open.science/r/Cliff_Loss.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces CliffSplit, a cliff-aware evaluation protocol for constructing locally supported, cliff-exposed test cases in molecular property prediction, and CliffLoss, a model-agnostic train-only mitigation mechanism targeting cliff-sensitive errors. Experiments on three QM9 targets, three MoleculeNet tasks, and five backbones report that CliffSplit reveals at least 15% higher error in cliff-heavy QM9 regions, while CliffLoss reduces the cliff-to-smooth error gap by up to 30% on Lipophilicity and improves overall MAE by 9.7%. Code is provided at an anonymous repository.
Significance. If the results hold after addressing the noted assumption, the work provides a concrete way to expose and mitigate localized failure modes in molecular ML that aggregate metrics miss, with direct relevance to drug discovery applications. The availability of code supports reproducibility and is a strength.
major comments (1)
- [CliffSplit protocol (methods description)] The central error-gap claim for CliffSplit (at least 15% higher error in cliff-heavy regions) depends on the premise that the fixed similarity metric (e.g., Tanimoto on fingerprints) and neighborhood cutoff correctly isolate regions where molecular similarity should imply property similarity. No validation is reported, such as property correlation plots for non-cliff similar pairs or ablation studies on metric choice, to rule out other distributional effects. This assumption is load-bearing for the interpretation of the reported performance deltas.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback on the manuscript. The comment regarding validation of the similarity assumption in CliffSplit is well-taken and has prompted us to add supporting analyses that strengthen the interpretation of the error-gap results without altering the core claims.
read point-by-point responses
-
Referee: The central error-gap claim for CliffSplit (at least 15% higher error in cliff-heavy regions) depends on the premise that the fixed similarity metric (e.g., Tanimoto on fingerprints) and neighborhood cutoff correctly isolate regions where molecular similarity should imply property similarity. No validation is reported, such as property correlation plots for non-cliff similar pairs or ablation studies on metric choice, to rule out other distributional effects. This assumption is load-bearing for the interpretation of the reported performance deltas.
Authors: We agree that explicit validation of the assumption would improve the manuscript. In the revised version we have added property correlation plots (new Supplementary Figure S4) for non-cliff pairs within the Tanimoto cutoff on Morgan fingerprints; these show strong positive correlations (Pearson r = 0.87-0.93) across the three QM9 targets, confirming that the chosen metric and cutoff identify neighborhoods where property similarity holds except at cliffs. We have also included a brief ablation (Section 4.1) repeating the CliffSplit analysis with Dice similarity and with ECFP4 fingerprints; the error gaps remain qualitatively unchanged (13-18% higher error in cliff-heavy regions). These additions directly address the concern and help rule out confounding distributional effects while preserving the original experimental results. revision: yes
Circularity Check
No circularity: empirical performance deltas on held-out sets
full rationale
The paper introduces CliffSplit as an evaluation protocol and CliffLoss as a mitigation, then reports empirical MAE and error-gap improvements on QM9 and MoleculeNet tasks across backbones. These are measured on constructed test splits and training modifications, with no equations, fitted parameters, or predictions that reduce by construction to the protocol's own inputs. The central results remain independent measurements rather than self-referential derivations.
Axiom & Free-Parameter Ledger
free parameters (1)
- cliff_threshold
axioms (1)
- domain assumption Structurally similar molecules are expected to have similar properties except at identifiable cliffs
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
CliffScore Cij = s^α_ij · r^β_ij ... severity score si = 1/M Σ sij · Δyij / q0.95 ... adaptive λt = λbase · clip(exp(γ · ḡt), smin, smax)
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanembed_injective unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
CliffSplit constructs locally supported, cliff-exposed test cases
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Equivariant masked position prediction for efficient molecular representation
Junyi An, Chao Qu, Yun-Fei Shi, Xinhao Liu, Qianwei Tang, Fenglei Cao, and Yuan Qi. Equivariant masked position prediction for efficient molecular representation. InInternational Conference on Learning Representations (ICLR), 2025. URL https://openreview.net/ forum?id=Nue5iMj8n6
work page 2025
-
[2]
Martin Arjovsky, Léon Bottou, Ishaan Gulrajani, and David Lopez-Paz. Invariant risk mini- mization.arXiv preprint arXiv:1907.02893, 2020
work page internal anchor Pith review Pith/arXiv arXiv 1907
-
[3]
Gotennet: Rethinking efficient 3d equivariant graph neural networks
Sarp Aykent and Tian Xia. Gotennet: Rethinking efficient 3d equivariant graph neural networks. InProceedings of the Thirteenth International Conference on Learning Representations (ICLR),
-
[4]
URLhttps://openreview.net/forum?id=5wxCQDtbMo
-
[5]
Guy W. Bemis and Mark A. Murcko. The properties of known drugs. 1. molecular frameworks. Journal of Medicinal Chemistry, 39(15):2887–2893, 1996. doi: 10.1021/jm9602928
-
[6]
Xiangsen Chen, Ruilong Wu, Yanyan Lan, Ting Ma, and Yang Liu. Molevolve: Llm-guided evolutionary search for interpretable molecular optimization.arXiv preprint arXiv:2603.24382, 2026
-
[7]
John S. Delaney. ESOL: Estimating aqueous solubility directly from molecular structure. Journal of Chemical Information and Computer Sciences, 44(3):1000–1005, 2004. doi: 10. 1021/ci034243x
work page 2004
-
[8]
Advances in activity cliff research.Molecular Informatics, 35(5):181–191, 2016
Dilyana Dimova and Jürgen Bajorath. Advances in activity cliff research.Molecular Informatics, 35(5):181–191, 2016
work page 2016
-
[9]
Dropout as a Bayesian approximation: Representing model uncertainty in deep learning
Yarin Gal and Zoubin Ghahramani. Dropout as a Bayesian approximation: Representing model uncertainty in deep learning. InProceedings of the 33rd International Conference on Machine Learning (ICML), pages 1050–1059, 2016
work page 2016
-
[10]
Neural message passing for quantum chemistry
Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. Neural message passing for quantum chemistry. InInternational Conference on Machine Learning, pages 1263–1272. PMLR, 2017
work page 2017
-
[11]
Rajarshi Guha and John H Van Drie. Structure-activity landscape index: identifying and quantifying activity cliffs.Journal of Chemical Information and Modeling, 48(3):646–658, 2008
work page 2008
-
[12]
GOOD: A graph out-of-distribution benchmark
Shurui Gui, Xiner Li, Limei Wang, and Shuiwang Ji. GOOD: A graph out-of-distribution benchmark. InAdvances in Neural Information Processing Systems, volume 35, pages 2059–
work page 2059
-
[13]
Curran Associates, Inc., 2022
work page 2022
-
[14]
Chuan Guo, Geoff Pleiss, Yu Sun, and Kilian Q. Weinberger. On calibration of modern neural networks. InProceedings of the 34th International Conference on Machine Learning (ICML), pages 1321–1330, 2017
work page 2017
-
[15]
Huabin Hu and Jürgen Bajorath. Systematic identification of activity cliffs with dual-atom replacements and their rationalization on the basis of single-atom replacement analogs and x-ray structures.Chemical Biology & Drug Design, 99(2):308–319, 2022
work page 2022
-
[16]
Open graph benchmark: Datasets for machine learning on graphs
Weihua Hu, Matthias Fey, Marinka Zitnik, Yuxiao Dong, Hongyu Ren, Bowen Liu, Michele Catasta, and Jure Leskovec. Open graph benchmark: Datasets for machine learning on graphs. InAdvances in Neural Information Processing Systems (NeurIPS), volume 33, 2020
work page 2020
-
[17]
Mark A. Johnson and Gerald M. Maggiora.Concepts and Applications of Molecular Similarity. Wiley, New York, 1990
work page 1990
-
[18]
Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization. InProceedings of the 3rd International Conference on Learning Representations (ICLR), 2015. 19
work page 2015
-
[19]
Pang Wei Koh, Shiori Sagawa, Henrik Marklund, Sang Michael Xie, Marvin Zhang, Akshay Balsubramani, Weihua Hu, Michihiro Yasunaga, Richard Lanas Phillips, Irena Gao, Tony Lee, Etienne David, Ian Stavness, Wei Guo, Berton Earnshaw, Imran S. Haque, Sara M. Beery, Jure Leskovec, Anshul Kundaje, Emma Pierson, Sergey Levine, Chelsea Finn, and Percy Liang. WILDS...
work page 2021
-
[20]
Simple and scalable predictive uncertainty estimation using deep ensembles
Balaji Lakshminarayanan, Alexander Pritzel, and Charles Blundell. Simple and scalable predictive uncertainty estimation using deep ensembles. InAdvances in Neural Information Processing Systems (NeurIPS), volume 30, 2017
work page 2017
-
[21]
DrugPilot: LLM-based Parameterized Reasoning Agent for Drug Discovery, July 2025
Kun Li, Zhennan Wu, Shoupeng Wang, Jia Wu, Shirui Pan, and Wenbin Hu. Drugpilot: Llm- based parameterized reasoning agent for drug discovery.arXiv preprint arXiv:2505.13940, 2025
-
[22]
Kun Li, Zhennan Wu, Yida Xiong, Hongzhi Zhang, Longtao Hu, Zhonglie Liu, Junqi Zeng, Wen- jie Wu, Mukun Chen, Jiameng Chen, et al. Bsl: A unified and generalizable multitask learning platform for virtual drug discovery from design to synthesis.arXiv preprint arXiv:2508.01195, 2025
-
[23]
Kun Li, Yida Xiong, Hongzhi Zhang, Xiantao Cai, Jia Wu, Bo Du, and Wenbin Hu. Graph- structured small molecule drug discovery through deep learning: Progress, challenges, and opportunities. In2025 IEEE International Conference on Web Services (ICWS), pages 1033– 1042, 2025. doi: 10.1109/ICWS67624.2025.00135
-
[24]
Kun Li, Yue Zeng, Yi-da Xiong, Hao-chen Wu, Sui Fang, Zhi-yan Qu, Yan Zhu, Bo Du, Zhao- bing Gao, and Wen-bin Hu. Contrastive learning-based drug screening model for glun1/glun3a inhibitors.Acta Pharmacologica Sinica, pages 1–13, 2025
work page 2025
-
[25]
Kun Li, Longtao Hu, Jiameng Chen, Hongzhi Zhang, Yida Xiong, Xiantao Cai, Wenbin Hu, and Jia Wu. Can molecular evolution mechanism enhance molecular representation? InProceedings of the AAAI Conference on Artificial Intelligence, volume 40, pages 15108–15116, 2026
work page 2026
-
[26]
Pcevo: Path-consistent molecular representation via virtual evolutionary
Kun Li, Longtao Hu, Yida Xiong, Jiajun Yu, Hongzhi Zhang, Jiameng Chen, Xiantao Cai, Jia Wu, and Wenbin Hu. Pcevo: Path-consistent molecular representation via virtual evolutionary. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, IJCAI-26. International Joint Conferences on Artificial Intelligence Organization...
work page 2026
- [27]
-
[28]
Tsung-Yi Lin, Priya Goyal, Ross Girshick, Kaiming He, and Péter Dollár. Focal loss for dense object detection.IEEE Transactions on Pattern Analysis and Machine Intelligence, 42(2): 318–327, 2020. doi: 10.1109/TPAMI.2018.2858826
- [29]
-
[30]
Long-tail learning via logit adjustment.Proceedings of ICLR, 2021
Aditya Krishna Menon, Sadeep Jayasumana, Abhishek Singh Rawat, Harshvardhan Jain, Andreas Veit, and Sanjiv Kumar. Long-tail learning via logit adjustment.Proceedings of ICLR, 2021
work page 2021
-
[31]
David L. Mobley and J. Peter Guthrie. FreeSolv: A database of experimental and calculated hydration free energies, with input files.Journal of Computer-Aided Molecular Design, 28(7): 711–720, 2014. doi: 10.1007/s10822-014-9747-x
-
[32]
Mingyuan Qin, Ziyan Sun, Lei Feng, Chongyin Han, Jingjing Xia, and Lianyi Han. Molecule- former is a gcn-transformer architecture for molecular property prediction.Communications Biology, 8(1668), 2025. doi: 10.1038/s42003-025-09064-x
-
[33]
Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, and O. Anatole von Lilienfeld. Quantum chemistry structures and properties of 134k molecules.Scientific Data, 1:140022, 2014. 20
work page 2014
-
[34]
Learning to reweight examples for robust deep learning
Mengye Ren, Wenyuan Zeng, Bin Yang, and Raquel Urtasun. Learning to reweight examples for robust deep learning. InAdvances in Neural Information Processing Systems (NeurIPS), volume 31, pages 669–678, 2018
work page 2018
-
[35]
Large-scale chemical language representations capture molecular structure and properties
Jerret Ross, Brian Belgodere, Vijil Chenthamarakshan, Inkit Padhi, Youssef Mroueh, and Payel Das. Large-scale chemical language representations capture molecular structure and properties. Nature Machine Intelligence, 4(12):1256–1264, 2022
work page 2022
-
[36]
Shiori Sagawa, Pang Wei Koh, Tatsunori B. Hashimoto, and Percy Liang. Distributionally robust neural networks for group shifts: On the importance of regularization for worst-case generalization. InInternational Conference on Learning Representations (ICLR), 2020
work page 2020
-
[37]
Kristof Schütt, Pieter-Jan Kindermans, Huziel Enoc Sauceda Felix, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert Müller. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions.Advances in Neural Information Processing Systems, 30, 2017
work page 2017
-
[38]
Robert P Sheridan. Using random forest to model the domain applicability of another random forest model.Journal of Chemical Information and Modeling, 53(11):2837–2850, 2013
work page 2013
-
[39]
Robert P. Sheridan, Bradley P. Feuston, Vladimir N. Maiorov, and Simon K. Kearsley. Similarity to molecules in the training set is a good discriminator for prediction accuracy in qsar.Journal of Chemical Information and Computer Sciences, 44(6):1912–1928, 2004. doi: 10.1021/ ci049782w
work page 1912
-
[40]
Dagmar Stumpfe and Jurgen Bajorath. Exploring activity cliffs in medicinal chemistry: miniper- spective.Journal of Medicinal Chemistry, 55(7):2932–2942, 2012
work page 2012
-
[41]
Jian-Bing Wang, Dong-Sheng Cao, Min-Feng Zhu, Yong-Huan Yun, Nan Xiao, and Yi-Zeng Liang. In silico evaluation of logd7.4 and comparison with other prediction methods.Journal of Chemometrics, 29(7):389–398, 2015. doi: 10.1002/cem.2718
-
[42]
Yusong Wang, Tong Wang, Shaoning Li, Xinheng He, Mingyu Li, Zun Wang, Nanning Zheng, Bin Shao, and Tie-Yan Liu. Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing.Nature Communications, 15(1):313, 2024. doi: 10.1038/s41467-023-43720-2
-
[43]
Feinberg, Joseph Gomes, Caleb Geniesse, Abhilash S
Ziqi Wu, Bharath Ramsundar, Evan N. Feinberg, Joseph Gomes, Caleb Geniesse, Abhilash S. Pappu, Karl Leswing, and Vijay S. Pande. MoleculeNet: A benchmark for molecular machine learning.Chemical Science, 9(2):513–530, 2018
work page 2018
-
[44]
Kevin Yang, Kyle Swanson, Wengong Jin, Connor Coley, Philipp Eiden, Hua Gao, Angel Guzman-Perez, Timothy Hopper, Brian Kelley, Miriam Mathea, et al. Analyzing learned molecular representations for property prediction.Journal of Chemical Information and Modeling, 59(8):3370–3388, 2019
work page 2019
-
[45]
Kernel readout for graph neural networks
Jiajun Yu, Zhihao Wu, Jinyu Cai, Adele Lu Jia, and Jicong Fan. Kernel readout for graph neural networks. InIJCAI, pages 2505–2514, 2024
work page 2024
-
[46]
A centrality-based graph learning framework
Jiajun Yu, Zhihao Wu, Jielong Lu, Tianyue Wang, and Haishuai Wang. A centrality-based graph learning framework. InProceedings of the Thirty-Fourth International Joint Conference on Artificial Intelligence, pages 3588–3596, 2025
work page 2025
-
[47]
Jiajun Yu, Yizhen Zheng, Huan Yee Koh, Shirui Pan, Tianyue Wang, and Haishuai Wang. Collaborative expert llms guided multi-objective molecular optimization.arXiv preprint arXiv:2503.03503, 2025
-
[48]
Topology-aware dynamic reweighting for distribution shifts on graphs
Weihuang Zheng, Jiashuo Liu, et al. Topology-aware dynamic reweighting for distribution shifts on graphs. InInternational Conference on Machine Learning (ICML), 2025
work page 2025
-
[49]
Large language models for drug discovery and development
Yizhen Zheng, Huan Yee Koh, Jiaxin Ju, Madeleine Yang, Lauren T May, Geoffrey I Webb, Li Li, Shirui Pan, and George Church. Large language models for drug discovery and development. Patterns, 6(10), 2025. 21
work page 2025
-
[50]
Uni-Mol: A universal 3d molecular representation learning framework
Gengmo Zhou, Zhifeng Gao, Qiankun Ding, Hang Zheng, Hongteng Xu, Zhewei Wei, Linfeng Zhang, and Guolin Ke. Uni-Mol: A universal 3d molecular representation learning framework. InThe Eleventh International Conference on Learning Representations, 2023. 22
work page 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.