Data-driven complete basis set limit estimates from a minimal auxiliary basis
Pith reviewed 2026-05-19 19:03 UTC · model grok-4.3
The pith
A pairwise interaction model plus minimal CABS baseline and kernel ridge regression estimates the complete basis set limit from a single minimal-basis calculation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the CBS energy can be estimated from a single quantum chemistry calculation in a minimal basis set by combining a pairwise interaction model with a minimal complementary auxiliary basis set baseline and applying a kernel ridge regression correction to the remaining error, which is more efficient than both direct and delta-machine learning, with the kernel models made tractable by Chebyshev polynomial approximations to atom-wise local kernels.
What carries the argument
Kernel ridge regression that corrects residuals after a pairwise interaction model augmented by a minimal CABS baseline, with atom-wise local kernels approximated via Chebyshev polynomials.
If this is right
- CBS energies become available after a single minimal-basis calculation rather than a series of increasing basis-set sizes.
- The physical baseline reduces the learning burden on the regression model, allowing smaller training sets to reach useful accuracy.
- Chebyshev approximations to local kernels lower the computational cost of training, making the method feasible on moderate hardware.
- The same hybrid baseline-plus-correction pattern can be applied to other slowly converging quantities in quantum chemistry.
Where Pith is reading between the lines
- The approach could be tested on larger or more diverse molecular datasets to check whether the baseline still leaves residuals that KRR can learn reliably.
- Similar physical baselines might accelerate convergence corrections for properties other than total energy, such as gradients or response functions.
- If the method scales, it would lower the barrier to obtaining CBS-quality data for high-throughput screening or machine-learning potentials.
Load-bearing premise
The pairwise interaction model and minimal CABS baseline together capture enough of the leading-order basis-set incompleteness effects that the KRR correction generalizes without large systematic residuals on new molecules.
What would settle it
Running explicit large-basis extrapolations on a held-out test set of molecules and finding that the KRR predictions show systematic errors larger than the improvement over the physical baseline alone would falsify the central claim.
Figures

read the original abstract
Quantum chemistry calculations are often performed using atom-centered basis sets which are chosen to balance accuracy and cost. While they are systematically improvable, the total energy converges slowly with basis set size towards the complete basis set (CBS) limit. Common extrapolation methods require several intermediate-quality calculations to afford an estimate of the CBS energy. We propose combining a pairwise interaction model with a minimal complementary auxiliary basis set (CABS) baseline to estimate the CBS energy from a single quantum chemistry calculation in a minimal basis set via Kernel-Ridge-Regression (KRR), which is more efficient than both direct and $\Delta$-machine learning. We show that KRR on standard molecular representations can be improved by approximating atom-wise local kernels using Chebyshev polynomials which allows us to train KRR models efficiently on moderate compute resources, further enabling a data-driven approach towards CBS combining physical baselines capturing leading order effects with data-efficient machine learning models.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes estimating the complete basis set (CBS) limit energy from a single minimal-basis quantum chemistry calculation by combining a pairwise interaction model with a minimal complementary auxiliary basis set (CABS) as a physical baseline, then applying Kernel Ridge Regression (KRR) on standard molecular representations (with Chebyshev-polynomial approximation of atom-wise local kernels) to correct residual basis-set incompleteness. The approach is positioned as more efficient than direct CBS extrapolation or Δ-machine learning.
Significance. If the numerical results hold, the work demonstrates a practical route to CBS-quality energies at the cost of one minimal-basis calculation by using a physically motivated baseline to capture leading-order incompleteness and a data-efficient ML correction for the remainder. The Chebyshev kernel approximation is a concrete technical contribution that enables training on moderate resources. The combination of explicit physical grounding with ML is a strength that could improve transferability over pure data-driven methods.
major comments (1)
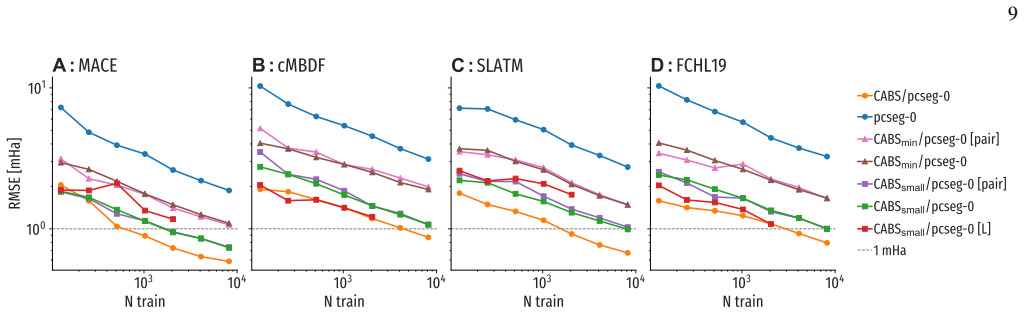
- [§4.3, Table 3] §4.3, Table 3: the reported residual MAE after the pairwise+CABS baseline is 0.08 kcal mol⁻¹ on the training distribution, but the corresponding value on the held-out set of larger molecules (n>20 atoms) rises to 0.22 kcal mol⁻¹; this directly tests whether the baseline leaves only a smooth local correction and therefore bears on the central generalization claim.
minor comments (3)
- [Eq. (7)] The definition of the minimal CABS in Eq. (7) uses the same symbol for the auxiliary functions as the standard CABS; a distinct subscript would remove ambiguity.
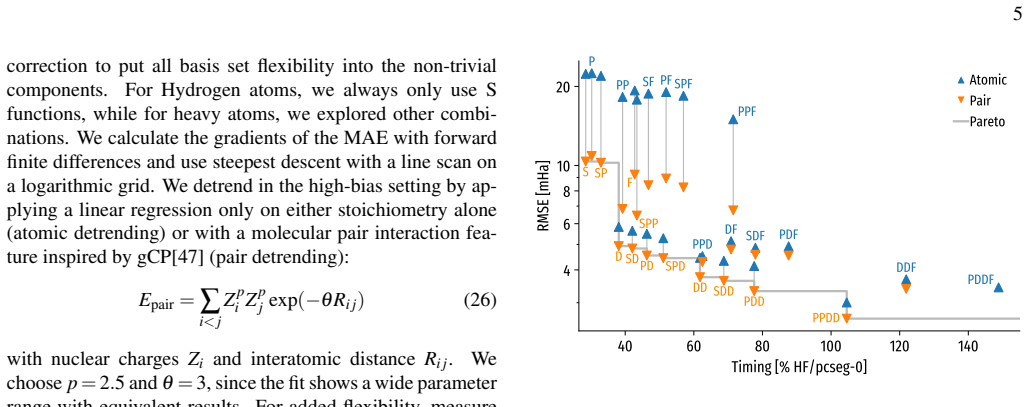
- [Figure 2] Figure 2 caption states 'learning curves for the KRR correction' but the y-axis label is missing the unit (kcal mol⁻¹); this affects readability of the data-efficiency claim.
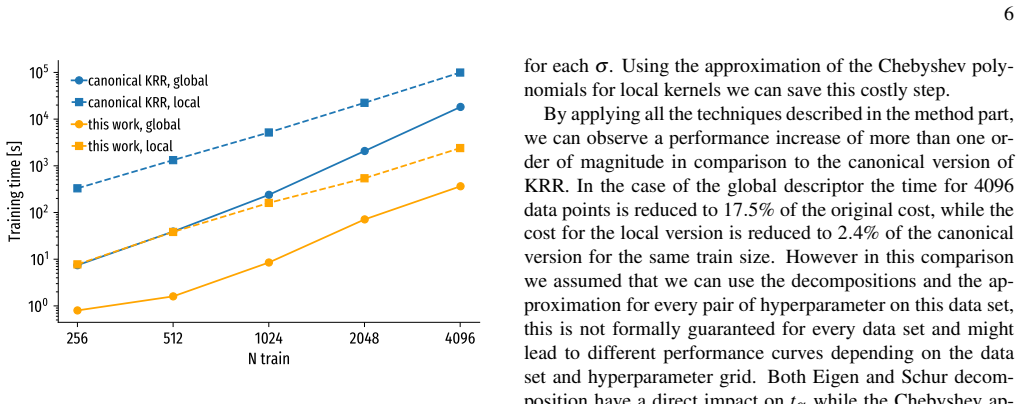
- [§5] The abstract claims the method is 'more efficient than both direct and Δ-machine learning' but provides no wall-time or scaling comparison; a brief statement in §5 would clarify the practical advantage.
Simulated Author's Rebuttal
We thank the referee for the constructive review and positive assessment of the significance of our work. We address the single major comment below and will revise the manuscript to strengthen the discussion of generalization.
read point-by-point responses
-
Referee: [§4.3, Table 3] §4.3, Table 3: the reported residual MAE after the pairwise+CABS baseline is 0.08 kcal mol⁻¹ on the training distribution, but the corresponding value on the held-out set of larger molecules (n>20 atoms) rises to 0.22 kcal mol⁻¹; this directly tests whether the baseline leaves only a smooth local correction and therefore bears on the central generalization claim.
Authors: We agree that the observed increase in residual MAE from 0.08 to 0.22 kcal mol⁻¹ on the held-out larger molecules is an important observation that directly relates to the generalization claim. This rise is expected given the greater number of pairwise interactions and potential for longer-range effects in systems with n>20 atoms, yet the absolute error remains chemically meaningful. To address this explicitly, we will revise §4.3 to include a plot of residual error versus number of atoms for the held-out set and add a short paragraph discussing the scaling of the local correction. This will clarify that the pairwise+CABS baseline successfully reduces the problem to a smooth, learnable residual even for larger molecules. revision: yes
Circularity Check
No circularity: physical baseline plus externally trained KRR correction remains independent of inputs.
full rationale
The claimed derivation rests on a pairwise interaction model combined with a minimal CABS baseline that supplies an independent physical approximation to leading basis-set incompleteness, followed by KRR trained on external quantum-chemistry data to learn residuals. Neither the baseline nor the learned correction reduces to the input minimal-basis energies by construction; the KRR step is a statistical fit to held-out higher-basis targets rather than a tautological re-expression of the training quantities. No self-citation chains, uniqueness theorems, or ansatz smuggling appear in the load-bearing steps. The method is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- KRR regularization and kernel hyperparameters
axioms (1)
- domain assumption Pairwise interaction model plus minimal CABS baseline captures leading-order basis-set incompleteness effects
Forward citations
Cited by 1 Pith paper
-
Property-Specific Molecular Representations via Feature-Space Transfer Compression
A transfer compression technique using semi-empirical data reduces molecular representation dimensions by a median 72% (range 36-98%) while retaining accuracy for energy, heat capacity, dipole moment and polarizabilit...
Reference graph
Works this paper leans on
-
[1]
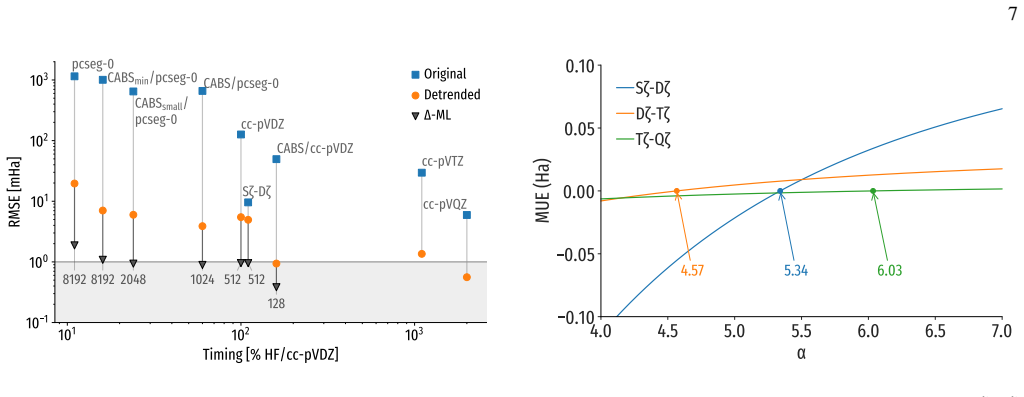
We estimated theαvalue that allows us to calculate the Sζ/Dζextrapolation employing the calculated RHF/pcseg-0 energies together with the RHF/cc-pVDZ retrieved from the dataset. Using 131554 data points available in the GDB- BSIE dataset, we obtained an optimalαvalue of 5.34 (Fig- ure 5). The calculation of theαvalue for the remaining cc- pVXZ family pair...
work page 2048
-
[2]
T. H. Dunning, Gaussian basis sets for use in correlated molecu- lar calculations. I. The atoms boron through neon and hydrogen, The Journal of Chemical Physics90, 1007 (1989)
work page 1989
-
[3]
T. Helgaker, W. Klopper, H. Koch, and J. Noga, Basis-set con- vergence of correlated calculations on water, The Journal of Chemical Physics106, 9639 (1997)
work page 1997
- [4]
-
[5]
A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen, and A. K. Wilson, Basis-set convergence in correlated calculations on Ne, N2, and H2O, Chemical Physics Letters 286, 243 (1998)
work page 1998
-
[6]
D. W. Schwenke, The extrapolation of one-electron basis sets in electronic structure calculations: How it should work and how it can be made to work, The Journal of Chemical Physics122, 014107 (2005)
work page 2005
-
[7]
A. M. Parameswaran, A. Fernández-Ramos, and D. G. Truhlar, Evaluating Cost and Accuracy in Two-Point Complete Basis Set Extrapolation Schemes Using Efficient Diffuse Basis Sets, The Journal of Physical Chemistry A128, 10673 (2024)
work page 2024
-
[8]
J. Lang, M. Przybytek, and M. Lesiuk, Estimating the Com- plete Basis Set Extrapolation Error through Random Walks, The Journal of Physical Chemistry Letters16, 4952 (2025). 10
work page 2025
-
[9]
J. M. L. Martin, Ab initio total atomization energies of small molecules — towards the basis set limit, Chemical Physics Let- ters259, 669 (1996)
work page 1996
-
[10]
D. G. Truhlar, Basis-set extrapolation, Chemical Physics Let- ters294, 45 (1998)
work page 1998
-
[11]
S. B. Huh and J. S. Lee, Basis set and correlation dependent extrapolation of correlation energy, The Journal of Chemical Physics118, 3035 (2003)
work page 2003
-
[12]
D. Bakowies, Extrapolation of electron correlation energies to finite and complete basis set targets, The Journal of Chemical Physics127, 084105 (2007)
work page 2007
-
[13]
D. Bakowies, Accurate extrapolation of electron correlation en- ergies from small basis sets, The Journal of Chemical Physics 127, 164109 (2007)
work page 2007
-
[14]
A. J. C. Varandas, Basis-set extrapolation of the correlation en- ergy, The Journal of Chemical Physics113, 8880 (2000)
work page 2000
-
[15]
W. Klopper and W. Kutzelnigg, Gaussian basis sets and the nuclear cusp problem, Journal of Molecular Structure: THEOCHEM135, 339 (1986)
work page 1986
-
[16]
D. Feller, Application of systematic sequences of wave func- tions to the water dimer, The Journal of Chemical Physics96, 6104 (1992)
work page 1992
-
[17]
F. Jensen, Polarization consistent basis sets: Principles, The Journal of Chemical Physics115, 9113 (2001)
work page 2001
-
[18]
Estimating the Hartree–Fock limit from finite basis set calculations
A. Karton and J. M. L. Martin, Comment on: “Estimating the Hartree–Fock limit from finite basis set calculations” [Jensen F (2005) Theor Chem Acc 113:267], Theoretical Chemistry Ac- counts115, 330 (2006)
work page 2005
-
[19]
G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley, and J. Mantzaris, A complete basis set model chemistry. I. The total energies of closed-shell atoms and hy- drides of the first-row elements, The Journal of Chemical Physics89, 2193 (1988)
work page 1988
-
[20]
J. M. L. Martin and P. R. Taylor, Benchmark quality total atom- ization energies of small polyatomic molecules, The Journal of Chemical Physics106, 8620 (1997)
work page 1997
-
[21]
F. Jensen, The basis set convergence of the Hartree–Fock en- ergy for H2, The Journal of Chemical Physics110, 6601 (1999)
work page 1999
-
[22]
F. Jensen, Estimating the Hartreeâ C”Fock limit from finite ba- sis set calculations, Theoretical Chemistry Accounts113, 267 (2005)
work page 2005
-
[23]
P. Kraus, Basis Set Extrapolations for Density Functional The- ory, Journal of Chemical Theory and Computation16, 5712 (2020)
work page 2020
-
[24]
F. Neese, The ORCA program system, Wiley Interdisciplinary Reviews: Computational Molecular Science2, 73 (2011)
work page 2011
-
[25]
K. A. Peterson, T. B. Adler, and H.-J. Werner, Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne, and Al–Ar, The Journal of Chemical Physics128, 084102 (2008)
work page 2008
-
[26]
T. B. Adler, G. Knizia, and H.-J. Werner, A simple and efficient CCSD(T)-F12 approximation, The Journal of Chemical Physics 127, 221106 (2007)
work page 2007
- [27]
- [28]
-
[29]
W. Kutzelnigg and W. Klopper, Wave functions with terms lin- ear in the interelectronic coordinates to take care of the corre- lation cusp. I. General theory, The Journal of Chemical Physics 94, 1985 (1991)
work page 1985
-
[30]
E. F. Valeev, Improving on the resolution of the identity in lin- ear R12 ab initio theories, Chemical Physics Letters395, 190 (2004)
work page 2004
-
[31]
R. A. Shaw and J. G. Hill, Approaching the Hartreeâ C“Fock Limit through the Complementary Auxiliary Basis Set Singles Correction and Auxiliary Basis Sets, Journal of Chemical The- ory and Computation13, 1691 (2017)
work page 2017
-
[32]
Liang and M. Head-Gordon, Approaching the Basis Set Limit in Density Functional Theory Calculations Using Dual Basis Sets without Diagonalization, The Journal of Physical Chem- istry A108, 3206 (2004)
work page 2004
-
[33]
K. Wolinski and P. Pulay, Second-order Møller–Plesset calcu- lations with dual basis sets, The Journal of Chemical Physics 118, 9497 (2003)
work page 2003
-
[34]
O. T. Unke and M. Meuwly, PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges, Journal of Chemical Theory and Computation15, 3678 (2019)
work page 2019
-
[35]
K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko, and K.-R. Müller, SchNet - a deep learning architecture for molecules and materials, The Journal of Chemical Physics148, 241722 (2018)
work page 2018
-
[36]
S. Holm, P. A. Unzueta, K. Thompson, and T. J. Martinez, Single-Point Extrapolation to the Complete Basis Set Limit through Deep Learning, Journal of Chemical Theory and Com- putation19, 4474 (2023)
work page 2023
-
[37]
D. T. Speckhard, C. Carbogno, L. M. Ghiringhelli, S. Lubeck, M. Scheffler, and C. Draxl, Extrapolation to the complete basis- set limit in density-functional theory using statistical learning, Physical Review Materials9, 013801 (2025)
work page 2025
-
[38]
Z. Qiao, M. Welborn, A. Anandkumar, F. R. Manby, and T. F. Miller, OrbNet: Deep Learning for Quantum Chemistry Us- ing Symmetry-Adapted Atomic-Orbital Features, The Journal of Chemical Physics153, 124111 (2020)
work page 2020
-
[39]
A. S. Christensen, S. K. Sirumalla, Z. Qiao, M. B. O’Connor, D. G. A. Smith, F. Ding, P. J. Bygrave, A. Anandkumar, M. Welborn, F. R. Manby, and T. F. Miller, OrbNet Denali: A machine learning potential for biological and organic chem- istry with semi-empirical cost and DFT accuracy, The Journal of Chemical Physics155, 204103 (2021)
work page 2021
-
[40]
Z. Qiao, A. S. Christensen, M. Welborn, F. R. Manby, A. Anandkumar, and T. F. Miller, Informing geometric deep learning with electronic interactions to accelerate quantum chemistry, Proceedings of the National Academy of Sciences 119, e2205221119 (2022)
work page 2022
-
[41]
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E(3)- equivariant graph neural networks for data-efficient and accu- rate interatomic potentials, Nature Communications13, 2453 (2022)
work page 2022
-
[42]
A. Otero-de-la Roza and G. A. DiLabio, Transferable Atom- Centered Potentials for the Correction of Basis Set Incomplete- ness Errors in Density-Functional Theory, Journal of Chemical Theory and Computation13, 3505 (2017)
work page 2017
-
[43]
A. Otero-de-la Roza and G. A. DiLabio, Improved Basis-Set In- completeness Potentials for Accurate Density-Functional The- ory Calculations in Large Systems, Journal of Chemical Theory and Computation16, 4176 (2020)
work page 2020
-
[44]
V . K. Prasad, A. Otero-de-la Roza, and G. A. DiLabio, Small- Basis Set Density-Functional Theory Methods Corrected with Atom-Centered Potentials, Journal of Chemical Theory and Computation18, 2913 (2022)
work page 2022
-
[45]
V . K. Prasad, A. Otero-de-la Roza, and G. A. DiLabio, Bridg- ing the gap between high-level quantum chemical methods and 11 deep learning models, Machine Learning: Science and Tech- nology5, 015035 (2024)
work page 2024
-
[46]
C. Qu, P. L. Houston, R. Conte, A. Nandi, and J. M. Bowman, Breaking the Coupled Cluster Barrier for Machine-Learned Po- tentials of Large Molecules: The Case of 15-Atom Acety- lacetone, The Journal of Physical Chemistry Letters12, 4902 (2021)
work page 2021
-
[47]
J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Dev- ereux, K. Barros, S. Tretiak, O. Isayev, and A. E. Roitberg, Ap- proaching coupled cluster accuracy with a general-purpose neu- ral network potential through transfer learning, Nature Commu- nications10, 10.1038/s41467-019-10827-4 (2019)
-
[48]
H. Kruse and S. Grimme, A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree- Fock and density functional theory calculations for large sys- tems, The Journal of Chemical Physics136, 154101 (2012)
work page 2012
- [49]
-
[50]
R. Ramakrishnan, P. O. Dral, M. Rupp, and O. A. von Lilien- feld, Big data meets quantum chemistry approximations: The Δ-machine learning approach, Journal of Chemical Theory and Computation11, 2087 (2015)
work page 2087
- [51]
- [52]
- [53]
-
[54]
W. J. Morgan, D. A. Matthews, M. Ringholm, J. Agarwal, J. Z. Gong, K. Ruud, W. D. Allen, J. F. Stanton, and H. F. Schaefer, Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde, Journal of Chemical Theory and Com- putation14, 1333 (2018)
work page 2018
-
[55]
A. Karton, Highly Accurate CCSDT(Q)/CBS Reaction Bar- rier Heights for a Diverse Set of Transition Structures: Basis Set Convergence and Cost-Effective Approaches for Estimating Post-CCSD(T) Contributions, The Journal of Physical Chem- istry A123, 6720 (2019)
work page 2019
-
[56]
G. Drabik and M. Rado ´n, Approaching the Complete Basis Set Limit for Spin-State Energetics of Mononuclear First-Row Transition Metal Complexes, Journal of Chemical Theory and Computation20, 3199 (2024)
work page 2024
- [57]
-
[58]
A. Ruiz-Serrano, N. D. M. Hine, and C.-K. Skylaris, Pulay forces from localized orbitals optimizedin situusing a psinc basis set, The Journal of Chemical Physics136, 234101 (2012)
work page 2012
-
[59]
M. F. Kasim, S. Lehtola, and S. M. Vinko, DQC: A Python pro- gram package for differentiable quantum chemistry, The Jour- nal of Chemical Physics156, 084801 (2022)
work page 2022
-
[60]
T. Tamayo-Mendoza, C. Kreisbeck, R. Lindh, and A. Aspuru- Guzik, Automatic differentiation in quantum chemistry with ap- plications to fully variational hartree–fock, ACS Central Sci- ence4, 559 (2018)
work page 2018
-
[61]
A. S. Abbott, B. Z. Abbott, J. M. Turney, and H. F. Schaefer, Arbitrary-order derivatives of quantum chemical methods via automatic differentiation, The Journal of Physical Chemistry Letters12, 3232 (2021)
work page 2021
-
[62]
X. Zhang and G. K.-L. Chan, Differentiable quantum chem- istry with PySCF for molecules and materials at the mean-field level and beyond, The Journal of Chemical Physics157, 204801 (2022)
work page 2022
-
[63]
G. Domenichini, G. F. von Rudorff, and O. A. von Lilienfeld, Effects of perturbation order and basis set on alchemical pre- dictions, The Journal of Chemical Physics153, 144118 (2020)
work page 2020
-
[64]
G. Domenichini, Extending the definition of atomic basis sets to atoms with fractional nuclear charge, The Journal of Chemical Physics160, 124107 (2024)
work page 2024
-
[65]
G. F. von Rudorff, Arbitrarily accurate quantum alchemy, The Journal of Chemical Physics , 224103 (2021)
work page 2021
-
[66]
O. A. von Lilienfeld, Accurate ab initio energy gradients in chemical compound space, Journal of Chemical Physics131, 164102 (2009)
work page 2009
-
[67]
A. Brakestad, S. R. Jensen, P. Wind, M. Dâ C™Alessandro, L. Genovese, K. H. Hopmann, and L. Frediani, Static Polar- izabilities at the Basis Set Limit: A Benchmark of 124 Species, Journal of Chemical Theory and Computation16, 4874 (2020)
work page 2020
-
[68]
A. Hurtado, H. Sekino, and R. J. Harrison, Benchmarking Correlation-Consistent Basis Sets for Frequency-Dependent Polarizabilities with Multiresolution Analysis, Journal of Chemical Theory and Computation20, 5145 (2024)
work page 2024
- [69]
-
[70]
G. F. von Rudorff, A. N. Artemyev, B. M. Lagutin, and P. V . De- mekhin, Optimal photoelectron circular dichroism of a model chiral system, The Journal of Chemical Physics160, 214301 (2024)
work page 2024
-
[71]
T.-T. Lu and S.-H. Shiou, Inverses of 2 × 2 block matrices, Computers & Mathematics with Applications43, 119 (2002)
work page 2002
-
[72]
B. Schölkopf, A. Smola, and K.-R. Müller, Nonlinear Compo- nent Analysis as a Kernel Eigenvalue Problem, Neural Compu- tation10, 1299 (1998)
work page 1998
-
[73]
F. A. Faber, A. S. Christensen, B. Huang, and O. A. von Lilien- feld, Alchemical and structural distribution based representa- tion for universal quantum machine learning, The Journal of Chemical Physics148, 241717 (2018)
work page 2018
-
[74]
E. C. Mitchell, J. M. Turney, and H. F. Schaefer, Automatic Dif- ferentiation for Explicitly Correlated MP2, Journal of Chemical Theory and Computation20, 8529 (2024)
work page 2024
-
[75]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K. Chan, PySCF: the Python-based simula- tions of chemistry framework (2017)
work page 2017
-
[76]
K. E. Yousaf and K. A. Peterson, Optimized auxiliary basis sets for explicitly correlated methods, The Journal of Chemi- cal Physics129, 184108 (2008)
work page 2008
-
[77]
J. G. Hill and K. A. Peterson, Correlation consistent basis sets for explicitly correlated wavefunctions: Pseudopotential-based basis sets for the post-dmain group elements Ga–Rn, The Jour- nal of Chemical Physics141, 094106 (2014)
work page 2014
-
[78]
E. Semidalas and J. M. L. Martin, Automatic generation of complementary auxiliary basis sets for explicitly correlated methods, Journal of Computational Chemistry43, 1690 (2022)
work page 2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.