k-Means Clustering in Fingerprint-Based Configuration Selection for Fitting Interatomic Potentials
Pith reviewed 2026-06-27 15:26 UTC · model grok-4.3
The pith
k-Means clustering on CrystalNN and RDF fingerprints selects smaller, more effective configuration sets for fitting EAM potentials than random selection.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper claims that k-means clustering applied to atomistic configuration fingerprints based on the CrystalNN model and radial distribution function improves the accuracy of fitting classical molecular dynamics interatomic potentials to density functional theory data for both energies and forces while requiring fewer configurations than random selection. For an EAM potential of titanium, this method achieves better precision and lower standard deviations, with only about 30 configurations sufficient to describe the full set of 1800 configurations well. The t-SNE reduction reveals overlaps between subsets with and without Ti vacancy, which k-means captures but random does not, and excluding
What carries the argument
k-means clustering on configuration fingerprints constructed from CrystalNN and RDF representations, which groups similar atomic environments to select diverse yet representative training configurations for potential fitting.
If this is right
- Only about 30 configurations suffice to obtain an EAM model that describes well the full set of 1800 configurations in terms of energies and forces.
- k-means clustering consistently achieves better precision and lower standard deviations for a smaller number of configurations than random selection.
- Overlapping configurations in t-SNE space indicate potential information redundancy that clustering can handle without loss of fit quality.
- When overlapping configurations with vacancies are excluded from the k-means selection and used only as a test set, their energy and force predictions show similar precision.
Where Pith is reading between the lines
- Similar fingerprint clustering could reduce the computational expense of generating large training sets for other interatomic potential forms or materials systems.
- The method highlights how dimensionality reduction techniques like t-SNE can help validate the completeness of selected configuration spaces.
- Applying this to larger or more complex datasets might reveal systematic redundancies in high-throughput simulation workflows.
Load-bearing premise
The fingerprints based on CrystalNN and RDF are assumed to capture the atomic-environment similarities that matter for energy and force accuracy in the subsequent EAM fit.
What would settle it
If a comparison on the same titanium DFT dataset shows that random selection of configurations achieves equal or better energy and force accuracy in EAM fits than the k-means selected sets of the same size, the advantage would be falsified.
Figures
read the original abstract
In this study, we present a method for selecting an arbitrary number of distinct configurations from a larger data set by applying k-means clustering to atomistic configuration fingerprints based on the CrystalNN model and radial distribution function (RDF). This approach improves the accuracy of fitting classical molecular dynamics interatomic potentials to density functional theory (DFT) data for both energies and forces while requiring fewer configurations than random selection. We demonstrate this improvement by fitting an embedded-atom method (EAM) potential for titanium, using various configurational sizes from an initial set of 1800 configurations. The k-means clustering consistently achieves better precision and lower standard deviations for a smaller number of configurations than random selection. The results also suggest that only about 30 configurations are sufficient to obtain an EAM model that describes well the full set of 1800 configurations in terms of energies and forces. Additionally, t-distributed stochastic neighbor embedding (t-SNE) method was used to reduce the configuration fingerprints into 2D space, and it revealed an overlap between two configuration subsets with and without Ti vacancy, indicating similar atomic environments. This similarity is captured by k-means clustering but not by random selection. Furthermore, when the overlapping configurations with vacancies were excluded from the k-means algorithm and used only as a test set, their energy and force predictions showed similar precision to those when they were included. This indicates that the overlapping configurations in the 2D t-SNE space indeed imply potential information redundancy among the atomistic configurations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes selecting training configurations for EAM potential fitting to Ti DFT data via k-means clustering on CrystalNN+RDF fingerprints. It claims this yields EAM models with better energy/force accuracy and lower variance than random selection, with ~30 configurations sufficing for the full 1800-configuration set; t-SNE visualization and a vacancy-exclusion test are used to argue that clustering captures redundancy.
Significance. If the fingerprint-to-property correlation holds, the method could reduce the DFT data volume required for interatomic potential development. The empirical comparison to random selection and the redundancy test via vacancy hold-out provide concrete support on this Ti dataset, though broader utility depends on the representation's fidelity.
major comments (3)
- [Results (comparison of k-means vs. random selection)] The central claim that k-means on CrystalNN+RDF fingerprints outperforms random selection for EAM accuracy rests on the untested assumption that Euclidean (or other) fingerprint distance tracks differences in per-atom energies and forces. No correlation analysis (e.g., rank correlation between fingerprint distances and |E_i - E_j| or force residuals on the held-out set) is reported; without it the reported improvement remains dataset-specific and does not explain why clustering should systematically beat random sampling.
- [Results (EAM fit accuracy vs. number of configurations)] The statement that 'only about 30 configurations are sufficient' and that k-means yields 'lower standard deviations' requires error-vs-N curves with error bars from repeated k-means initializations and random draws, plus a statistical test (e.g., paired t-test or Wilcoxon) on the RMSE distributions. The abstract supplies no numerical RMSE values or p-values, so the precision and variance claims cannot be evaluated for robustness.
- [t-SNE analysis and vacancy test] t-SNE is used to demonstrate overlap between vacancy and non-vacancy configurations, yet t-SNE is a nonlinear embedding that does not preserve original distances; therefore overlap in the 2-D projection does not confirm that the high-dimensional fingerprints used by k-means actually group configurations with similar EAM energies/forces. A direct check in the original fingerprint space (e.g., intra- vs. inter-cluster distance statistics) is needed.
minor comments (2)
- [Methods] The EAM fitting protocol (loss-function weights between energy and force terms, optimizer, convergence criteria, and how the 'full set of 1800 configurations' is used for validation) should be stated explicitly so that the reported precision can be reproduced.
- [Figures] Figure captions and axis labels for the error-vs-N plots should include the exact definition of the error metric (RMSE per atom? total energy?) and the number of independent trials used for the standard-deviation shading.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments. We address each major point below and indicate the revisions we will make to strengthen the manuscript.
read point-by-point responses
-
Referee: [Results (comparison of k-means vs. random selection)] The central claim that k-means on CrystalNN+RDF fingerprints outperforms random selection for EAM accuracy rests on the untested assumption that Euclidean (or other) fingerprint distance tracks differences in per-atom energies and forces. No correlation analysis (e.g., rank correlation between fingerprint distances and |E_i - E_j| or force residuals on the held-out set) is reported; without it the reported improvement remains dataset-specific and does not explain why clustering should systematically beat random sampling.
Authors: We agree that an explicit correlation analysis would provide stronger justification for why the clustering approach outperforms random selection. Our current work presents an empirical demonstration on the Ti dataset, where the method yields measurable improvements in EAM accuracy. To address the concern, we will add a rank-correlation analysis (e.g., Spearman) between fingerprint distances and energy/force differences on held-out configurations in the revised manuscript. revision: yes
-
Referee: [Results (EAM fit accuracy vs. number of configurations)] The statement that 'only about 30 configurations are sufficient' and that k-means yields 'lower standard deviations' requires error-vs-N curves with error bars from repeated k-means initializations and random draws, plus a statistical test (e.g., paired t-test or Wilcoxon) on the RMSE distributions. The abstract supplies no numerical RMSE values or p-values, so the precision and variance claims cannot be evaluated for robustness.
Authors: We accept that the robustness of the claims would benefit from the requested statistical elements. In the revision we will include error-versus-N curves with error bars obtained from multiple k-means initializations and random draws, report explicit RMSE values, apply a statistical test such as the Wilcoxon signed-rank test to the RMSE distributions, and update the abstract with numerical results and significance values. revision: yes
-
Referee: [t-SNE analysis and vacancy test] t-SNE is used to demonstrate overlap between vacancy and non-vacancy configurations, yet t-SNE is a nonlinear embedding that does not preserve original distances; therefore overlap in the 2-D projection does not confirm that the high-dimensional fingerprints used by k-means actually group configurations with similar EAM energies/forces. A direct check in the original fingerprint space (e.g., intra- vs. inter-cluster distance statistics) is needed.
Authors: We recognize that t-SNE is a visualization tool that does not preserve distances and therefore cannot alone confirm clustering behavior in the original space. We will add a direct analysis in the high-dimensional fingerprint space, including intra- versus inter-cluster distance statistics, to corroborate that the k-means groupings correspond to similar atomic environments relevant to the EAM fit. revision: yes
Circularity Check
No circularity: empirical comparison to random baseline is independent of clustering definition
full rationale
The paper reports an empirical result: k-means on CrystalNN+RDF fingerprints produces configuration subsets that yield lower EAM energy/force errors (and lower variance) than random subsets of the same size when fitted to the 1800-configuration Ti DFT set. This is measured directly on held-out or full-set residuals after fitting; the improvement is not defined into existence by the clustering algorithm or any fitted parameter. The t-SNE overlap test is a post-hoc visualization confirming redundancy capture, not a derivation step. No equations, self-citations, or ansatzes reduce the central claim to a tautology with the input fingerprints or selection procedure. The method is externally falsifiable against the random baseline on the same data.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption k-means clustering applied to the chosen fingerprints will produce representative subsets for EAM fitting
Reference graph
Works this paper leans on
-
[1]
This selection should aim to minimize the training dataset size while maximizing the representation of the underlying potential energy surface (PES)
Introduction In the development of classical or machine-learning interatomic potentials based on ab initio density functional theory (DFT) data, an important task is the selection of suitable atomistic configurations for fitting or learning 1–3. This selection should aim to minimize the training dataset size while maximizing the representation of the unde...
-
[2]
Methodology 2.1 Configuration Fingerprint and K-means Clustering The fingerprint characterizing each atomistic configuration was defined as consisting of three parts:
-
[3]
These descriptors quantify the likelihood of different coordination numbers and the resemblance to specific geometric configurations
CrystalNN (244 elements, for geometrical atomic environments) The CrystalNN method generates a 61-dimensional vector for each atom within a configuration, capturing various aspects of its local atomic coordination environment using coordination descriptors. These descriptors quantify the likelihood of different coordination numbers and the resemblance to ...
-
[4]
The values are averaged over each atomic site, resulting in four metrics: mean, standard, minimum, and maximum value
Atomic mass difference (4 elements , for differences in local element composition) This part represents the aggregate differences in atomic masses between an atom and its neighbors (up to 10 Å) in the configuration. The values are averaged over each atomic site, resulting in four metrics: mean, standard, minimum, and maximum value
-
[5]
Average bond distance (4 elements) , radial distribution function (100 elements, for different distances between atoms / lattice parameters) The first 4 elements represent statistical measures (mean, standard deviation, minimum and maximum) of the average bond distances for each atomic site and its neighboring pairs (up to 10 Å) within a configuration. Th...
-
[6]
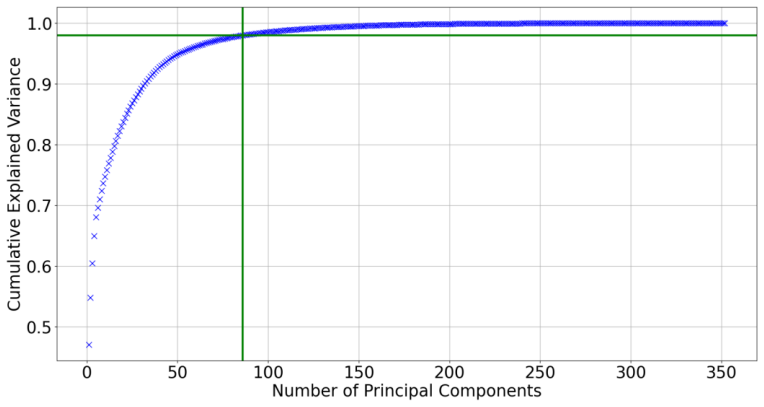
The first 85 components accounted for a cumulative explained variance of 98% (Fig
Results 3.1 Clustering of Ti Configurations The 352-element fingerprints of the generated Ti set, consisting of 1800 configurations, were analyzed using PCA. The first 85 components accounted for a cumulative explained variance of 98% (Fig. 2). This number of components was used for each configuration as its reduced fingerprint, and they were clustered us...
-
[7]
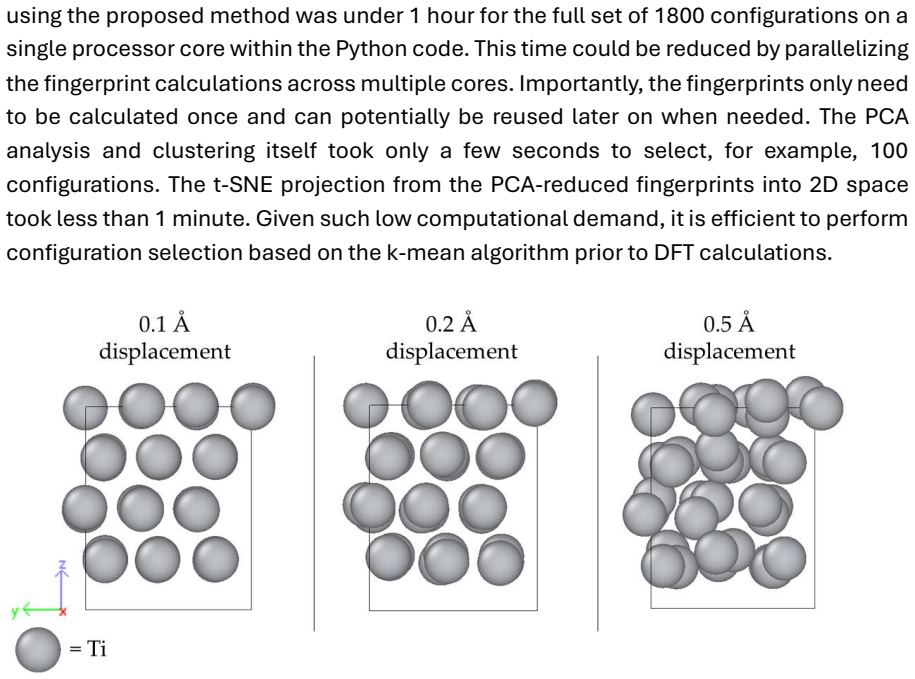
One group includes the supercells without vacancies and with a maximum atomic displacement of up to 0.1 Å (marked as 'Standard 0.1')
-
[8]
The second group consists of supercells with no vacancy and displacements of up to 0.2 Å ('Standard 0.2')
-
[9]
The third group features supercells with one Ti vacancy and a maximum displacement of 0.1 Å ('Vaca 0.1')
-
[10]
The fourth group contains supercells with one Ti vacancy and a maximum displacement of 0.2 Å ('Vaca 0.2')
-
[11]
The fifth group is formed by supercells without vacancies and with a maximum displacement of 0.5 Å ('Standard 0.5'), as well as supercells with one Ti vacancy and a maximum displacement of 0.5 Å ('Vaca 0.5'). The fifth group exhibits considerable overlap between non -vacancy and vacancy Ti supercells, indicating redundancy (i.e., similarity in atomic envi...
-
[12]
The advantages of this selection method were shown in the fitting of the EAM potential for Ti with an initial configuration size of 1800
Conclusions This study demonstrates the effectiveness of using k -means clustering based on atomistic configuration fingerprints combining CrystalNN and RDF to select configurations from a larger generated set for fitting MD interatomic potentials on DFT data. The advantages of this selection method were shown in the fitting of the EAM potential for Ti wi...
-
[13]
SGS24/121/OHK2/3T/12]
Acknowledgments This work was supported by the Grant Agency of the Czech Technical University in Prague [grant No. SGS24/121/OHK2/3T/12]. Computational resources were provided by the e - INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic. I would also like to thank Pavel Baláž for his valuable advic...
-
[14]
Machine Learning Interatomic Potentials: Keys to First-Principles Multiscale Modeling
References (1) Mortazavi, B. Machine Learning Interatomic Potentials: Keys to First-Principles Multiscale Modeling. In Machine Learning in Modeling and Simulation: Methods and Applications; Springer, 2023; pp 427–451. (2) Eyert, V .; Wormald, J.; Curtin, W. A.; Wimmer, E. Machine-Learned Interatomic Potentials: Recent Developments and Prospective Applicat...
arXiv 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.