Intrinsic Defect Energetics and Fluorine Doping Effects in Li2CO3 and Li2O2: A First-Principles Study
Pith reviewed 2026-06-25 21:05 UTC · model grok-4.3
The pith
Fluorine doping lowers lithium and carbon vacancy formation energies selectively in Li2CO3 and lithium vacancy energy in Li2O2
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
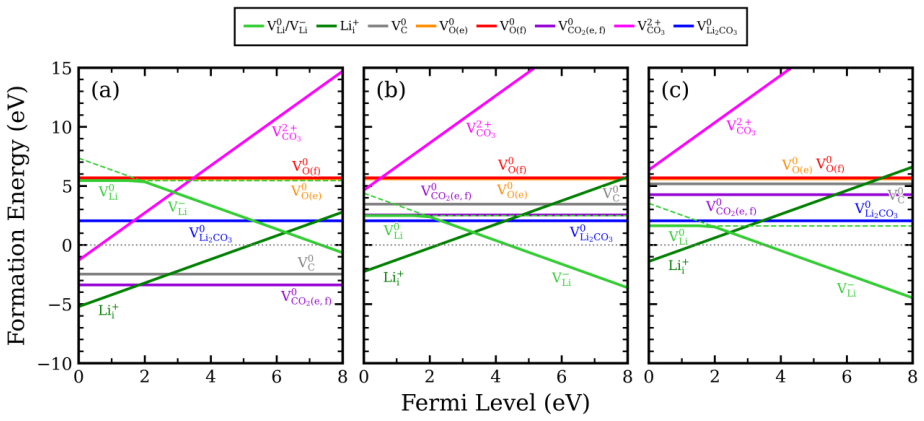
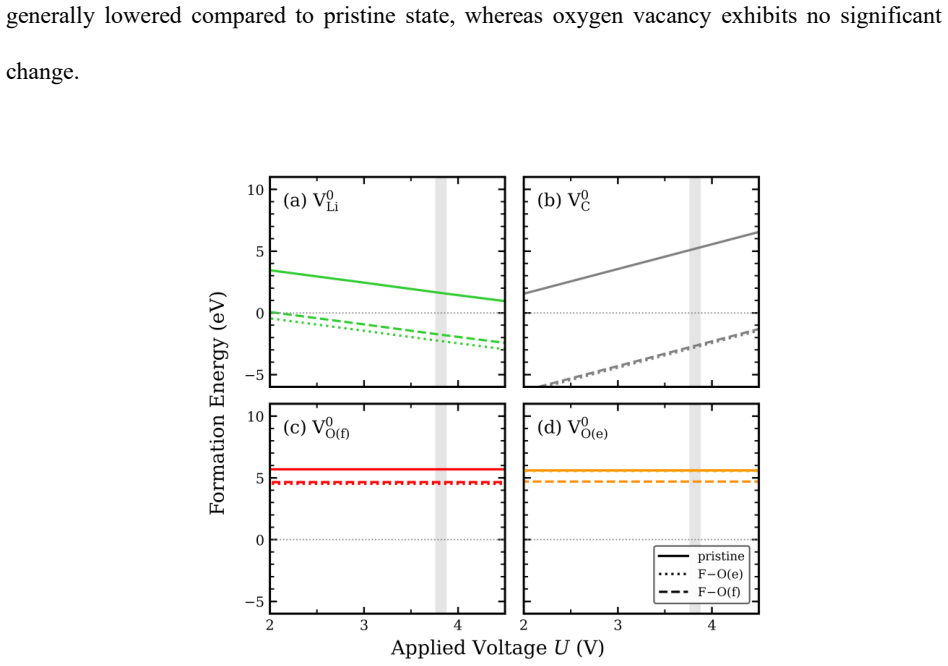
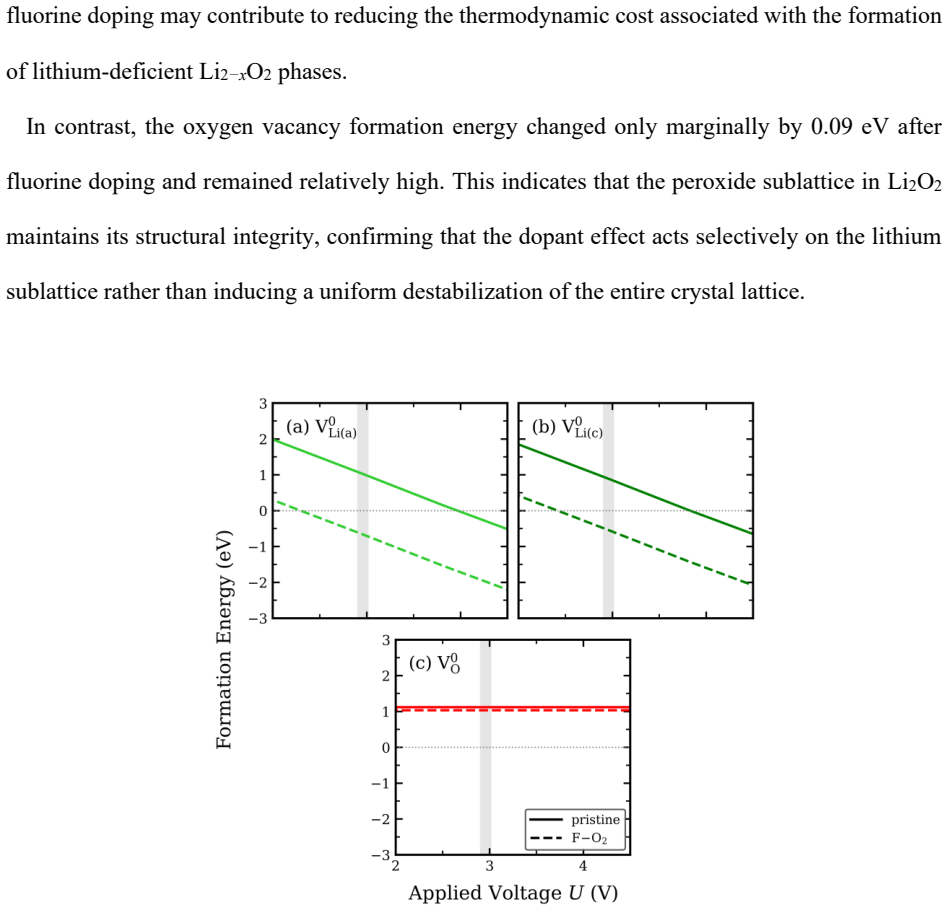
Intrinsic defect analysis reveals that defect behavior is predominantly governed by lithium-related defects. Upon fluorine doping, lithium and carbon vacancy formation energies decrease selectively in Li2CO3, partially destabilizing the carbonate framework, while a reduction in lithium vacancy formation energy is also observed in Li2O2. These results suggest that fluorine doping modulates the defect energetics of both discharge products, potentially providing a thermodynamic basis for controlling the stability of Li2CO3 and Li2O2 under thermodynamic conditions representative of lithium-oxygen batteries.
What carries the argument
First-principles calculations of vacancy formation energies before and after fluorine substitution, performed in supercells with chosen chemical potentials
If this is right
- Lithium and carbon vacancy formation energies decrease selectively in Li2CO3 upon fluorine doping.
- Lithium vacancy formation energy also decreases in Li2O2.
- The carbonate framework experiences partial destabilization in Li2CO3.
- Doping provides a thermodynamic route to influence stability of both discharge products.
Where Pith is reading between the lines
- The selective energy reductions could alter relative decomposition rates of carbonate versus peroxide phases during cell operation.
- Similar doping approaches might be explored in related alkali metal compounds to tune defect-controlled properties.
- Validation through direct defect spectroscopy on doped samples would clarify applicability beyond the computed conditions.
Load-bearing premise
The first-principles defect energies computed under the chosen supercell and chemical-potential conditions accurately reflect thermodynamic stability under the operating conditions of lithium-oxygen batteries.
What would settle it
Experimental measurement of lithium or carbon vacancy concentrations in fluorine-doped Li2CO3 and Li2O2 samples at battery-relevant chemical potentials would directly test whether the computed energy reductions occur.
Figures
read the original abstract
Lithium carbonate, Li2CO3, is a thermodynamically stable carbonate phase whose defect energetics are closely related to its stability and decomposition behavior in various lithium-based electrochemical systems. These properties of Li2CO3 are particularly important in lithium-oxygen battery environments. In these systems, Li2CO3 can form as a parasitic discharge product alongside Li2O2, the primary discharge product, leading to performance degradation. However, compared with Li2O2, the intrinsic defect thermodynamics of Li2CO3 and how chemical doping modifies its defect energetics remain insufficiently understood. In this study, first-principles calculations were performed to systematically analyze the intrinsic point-defect energetics of Li2CO3 and to evaluate the effects of fluorine doping on vacancy formation energies in Li2CO3 and Li2O2. Intrinsic defect analysis reveals that defect behavior is predominantly governed by lithium-related defects. Upon fluorine doping, lithium and carbon vacancy formation energies decrease selectively in Li2CO3, partially destabilizing the carbonate framework, while a reduction in lithium vacancy formation energy is also observed in Li2O2. These results suggest that fluorine doping modulates the defect energetics of both discharge products, potentially providing a thermodynamic basis for controlling the stability of Li2CO3 and Li2O2 under thermodynamic conditions representative of lithium-oxygen batteries.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports first-principles DFT calculations of intrinsic point-defect energetics in Li2CO3 and Li2O2, with emphasis on vacancy formation energies, and evaluates the effects of fluorine doping on these energies. The central claims are that defect behavior is dominated by lithium-related defects, that F doping selectively lowers Li and C vacancy formation energies in Li2CO3 (partially destabilizing the carbonate), and that Li vacancy formation energy is also reduced in Li2O2, all under thermodynamic conditions stated to be representative of lithium-oxygen batteries.
Significance. If the computed formation energies remain representative when referenced to the electrochemical potentials set by Li-O2 cell operation, the results could supply a thermodynamic rationale for using F doping to modulate stability of parasitic Li2CO3 versus the desired Li2O2 product. The work employs standard supercell defect methodology and formation-energy formulas; its value would be strengthened by explicit validation or sensitivity checks against experimental defect data or voltage-dependent conditions.
major comments (1)
- [Abstract] Abstract and (presumed) Methods/Results sections: the claim that calculations are performed “under thermodynamic conditions representative of lithium-oxygen batteries” is load-bearing for the interpretation that the reported decreases in vacancy energies imply destabilization under operating conditions. No explicit justification, chemical-potential references (Li, C, O, F), finite-size corrections, or sensitivity analysis to the ~2.96 V cell voltage is supplied in the provided text; if the potentials are instead taken from elemental phases or the Li2CO3/Li2O2 equilibrium without voltage adjustment, the selective lowering may not translate to the battery regime.
minor comments (1)
- [Abstract] The abstract supplies no numerical formation-energy values, supercell sizes, or error estimates; inclusion of at least representative numbers and a brief statement of convergence would improve immediate assessability.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address the single major comment below and will revise the manuscript accordingly to strengthen the presentation of thermodynamic conditions.
read point-by-point responses
-
Referee: [Abstract] Abstract and (presumed) Methods/Results sections: the claim that calculations are performed “under thermodynamic conditions representative of lithium-oxygen batteries” is load-bearing for the interpretation that the reported decreases in vacancy energies imply destabilization under operating conditions. No explicit justification, chemical-potential references (Li, C, O, F), finite-size corrections, or sensitivity analysis to the ~2.96 V cell voltage is supplied in the provided text; if the potentials are instead taken from elemental phases or the Li2CO3/Li2O2 equilibrium without voltage adjustment, the selective lowering may not translate to the battery regime.
Authors: We agree that explicit details on chemical-potential references, finite-size corrections, and relation to cell voltage would improve clarity. The calculations reference Li to bcc-Li, C to graphite, O to O2(g), and F to F2(g) at standard states, with formation energies computed via the standard supercell formula; finite-size corrections follow the FNV scheme. The doping-induced reductions in vacancy energies are relative quantities and thus independent of the absolute reference; these trends remain informative for battery conditions even if absolute values shift with voltage. We will add an explicit subsection in Methods/Results detailing the references, confirming FNV corrections were used, and providing a short discussion of how the reported trends map onto the ~2.96 V Li-O2 equilibrium (including a one-paragraph sensitivity note). revision: yes
Circularity Check
No circularity detected; derivation is self-contained first-principles computation
full rationale
The paper computes intrinsic point-defect formation energies and fluorine-doping effects in Li2CO3 and Li2O2 via standard DFT supercell calculations and the conventional defect formation-energy formula. No fitted parameters are renamed as predictions, no self-definitional loops appear in the energy expressions, and no load-bearing uniqueness theorems or ansatzes are imported from the authors' prior work. The central results follow directly from the electronic-structure calculations under stated chemical-potential references; the mapping to battery conditions is an interpretive step external to the numerical derivation itself.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard density-functional-theory approximations and supercell boundary conditions suffice to compute formation energies of point defects in these ionic solids.
Reference graph
Works this paper leans on
-
[1]
D.; Siegel, D
(1) Radin, M. D.; Siegel, D. J. Charge Transport in Lithium Peroxide: Relevance for Rechargeable Metal –Air Batteries. Energy Environ. Sci. 2013, 6 (8),
2013
-
[2]
https://doi.org/10.1039/c3ee41632a. (2) Cortes, H. A.; Vildosola, V. L.; Barral, M. A.; Corti, H. R. Effect of Halogen Dopants on the Properties of Li 2 O2 : Is Chloride Special? Phys. Chem. Chem. Phys. 2018, 20 (25), 16924– 16931. https://doi.org/10.1039/C8CP01211C. (3) Thomas C. Allison. NIST -JANAF Thermochemical Tables - SRD 13,
-
[3]
(4) Idemoto, Y.; Richardson, J
https://doi.org/10.18434/T42S31. (4) Idemoto, Y.; Richardson, J. W.; Koura, N.; Kohara, S.; Loong, C. -K. Crystal Structure of (LixK1 − x)2CO3 (x = 0, 0.43, 0.5, 0.62,
-
[4]
Journal of Physics and Chemistry of Solids 1998, 59 (3), 363 –376
by Neutron Powder Diffraction Analysis. Journal of Physics and Chemistry of Solids 1998, 59 (3), 363 –376. https://doi.org/10.1016/S0022 - 3697(97)00209-6
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.