Reaching the thermodynamic limit of periodic CCSD cohesive energies and band gaps with denser Brillouin zone sampling
Pith reviewed 2026-06-27 05:41 UTC · model grok-4.3
The pith
A distributed-memory periodic CCSD implementation enables 216 k-point sampling to converge cohesive energies and band gaps to 0.1 eV.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the new distributed-memory software implementation of periodic CCSD runs efficiently on up to 12 nodes, allowing calculations in which the Brillouin zone is sampled with up to 6^3=216 k-points. This enables reliable extrapolation to the thermodynamic limit. For eight simple semiconductors and insulators, the cohesive energy and band gap are reported converged to 0.1 eV, providing definitive benchmark numbers for the CCSD level of theory.
What carries the argument
Distributed-memory implementation of periodic coupled-cluster theory with single and double excitations (CCSD) that supports dense Brillouin zone sampling up to 216 k-points for thermodynamic limit extrapolation.
If this is right
- The reported values serve as benchmarks for assessing other electronic structure methods in periodic systems.
- Cohesive energies at the CCSD level typically underestimate experimental values by 0.1-0.2 eV on average.
- Band gaps at the CCSD level typically overestimate experimental values by about 0.4 eV on average.
- Ground-state and excited-state properties in solids can now be computed with higher k-point density before extrapolation.
Where Pith is reading between the lines
- The benchmarks could be used to test and improve lower-cost methods such as density functional theory for solid-state properties.
- The distributed implementation approach may extend to higher-order coupled-cluster methods or larger unit cells as hardware improves.
- Similar dense sampling strategies could reduce finite-size errors in other many-body perturbation theories for solids.
Load-bearing premise
The new distributed-memory implementation correctly computes the periodic CCSD energies without numerical errors, and the extrapolation procedure from finite k-point samplings accurately reaches the thermodynamic limit.
What would settle it
A calculation with substantially denser k-point sampling such as 8^3=512 points that deviates by more than 0.1 eV from the reported extrapolated values would falsify the convergence claim.
Figures
read the original abstract
The high computational cost of periodic coupled-cluster theory has limited the density of Brillouin zone sampling, yielding finite-size errors that need to be removed by extrapolation. Here we report the development and application of a distributed-memory software implementation of periodic coupled-cluster theory with single and double excitations (CCSD) that runs efficiently on up to 12 nodes with 96 cores each. This new implementation allows ground-state and excited-state calculations in which the Brillouin zone is sampled with up to $6^3=216$ $k$-points, allowing us to reliably extrapolate to the thermodynamic limit. For eight simple semiconductors and insulators, we report the cohesive energy and band gap, which are converged to 0.1 eV, providing definitive benchmark numbers for the CCSD level of theory. Compared to experimental values, average errors for the cohesive energy are 0.1-0.2 eV (typically an underestimate), and average errors for the band gap are about 0.4 eV (typically an overestimate).
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports the development of a distributed-memory implementation of periodic CCSD that enables Brillouin zone sampling with up to 6^3 k-points. For eight simple semiconductors and insulators, it provides cohesive energies and band gaps extrapolated to the thermodynamic limit, claimed to converge to 0.1 eV, serving as benchmarks, with average errors versus experiment of 0.1-0.2 eV (cohesive energies) and ~0.4 eV (band gaps).
Significance. If the implementation is numerically correct and the extrapolation reliable, the work would supply useful high-accuracy periodic CCSD benchmarks that address finite-size errors limiting prior studies; the technical advance in scaling to denser k-grids is a clear strength.
major comments (2)
- [Implementation section] Implementation section: no comparisons are provided between the new distributed-memory periodic CCSD results at small k-grids (e.g., 2^3 or 3^3) and existing periodic CCSD codes, leaving the numerical correctness of the energies unverified and directly undermining the 0.1 eV convergence claim.
- [Results/extrapolation section] Results/extrapolation section: the manuscript provides no explicit description of the extrapolation functional form, fitting procedure, or quantitative error analysis demonstrating that finite-size errors have been removed to the stated 0.1 eV precision for the reported cohesive energies and band gaps.
minor comments (1)
- [Abstract] Abstract: the phrasing 'definitive benchmark numbers' is not yet supported by the validation data shown.
Simulated Author's Rebuttal
We thank the referee for their constructive comments on our manuscript. We address each major comment below and will revise the manuscript accordingly to strengthen the presentation of our results.
read point-by-point responses
-
Referee: [Implementation section] Implementation section: no comparisons are provided between the new distributed-memory periodic CCSD results at small k-grids (e.g., 2^3 or 3^3) and existing periodic CCSD codes, leaving the numerical correctness of the energies unverified and directly undermining the 0.1 eV convergence claim.
Authors: We agree that explicit numerical verification against existing periodic CCSD implementations is important for establishing correctness. In the revised manuscript, we will add direct comparisons of our distributed-memory CCSD energies at 2^3 and 3^3 k-grids for representative systems against published results from other periodic CCSD codes. These comparisons will be included in the Implementation section to support the reliability of the implementation and the subsequent convergence claims. revision: yes
-
Referee: [Results/extrapolation section] Results/extrapolation section: the manuscript provides no explicit description of the extrapolation functional form, fitting procedure, or quantitative error analysis demonstrating that finite-size errors have been removed to the stated 0.1 eV precision for the reported cohesive energies and band gaps.
Authors: We acknowledge that the extrapolation details require clarification. The revised manuscript will explicitly describe the functional form employed for extrapolation to the thermodynamic limit (typically of the form a + b/N_k + c/N_k^{4/3} or similar, as appropriate for the quantity), the fitting procedure used, and quantitative error estimates (e.g., standard errors from the fit and sensitivity to the number of k-points included). This will demonstrate that residual finite-size errors are below the stated 0.1 eV threshold for the reported cohesive energies and band gaps. revision: yes
Circularity Check
No circularity; results from direct computation on new implementation with standard extrapolation
full rationale
The paper reports development of a distributed-memory periodic CCSD code and its application to compute cohesive energies and band gaps at up to 6^3 k-points for eight materials, followed by extrapolation to the thermodynamic limit. No load-bearing step reduces by construction to a fitted parameter, self-definition, or self-citation chain. The extrapolation is presented as a numerical procedure to remove finite-size errors, not as a prediction derived from the same data by definition. Claims rest on the correctness of the implementation and the validity of the extrapolation form, which are external to any circular reduction within the paper's equations. This is a standard computational reporting structure with independent content.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
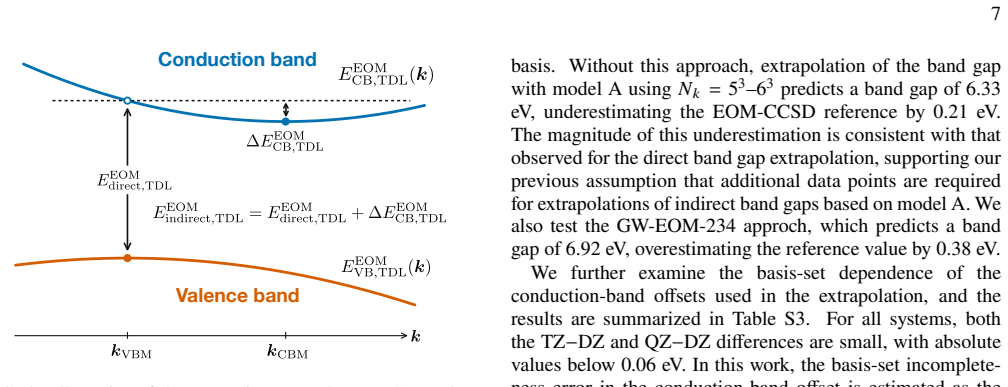
The band gap is then obtained as the sum of the IP and EA
−𝐸(𝑁)and𝐸(𝑁+1) −𝐸(𝑁), respectively, where𝐸(𝑁)is the ground-state energy of an𝑁-electron system. The band gap is then obtained as the sum of the IP and EA. When necessary, IP and EA calculations are performed using shifted𝑘-meshes to include the𝑘-points corresponding to the valence band maximum (VBM) or conduction band minimum (CBM). The 𝑘-points correspon...
-
[2]
These results are summarized in Table III
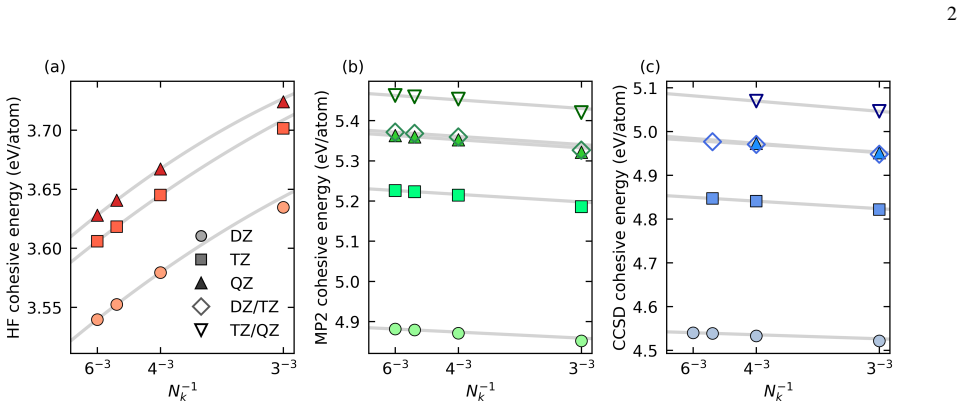
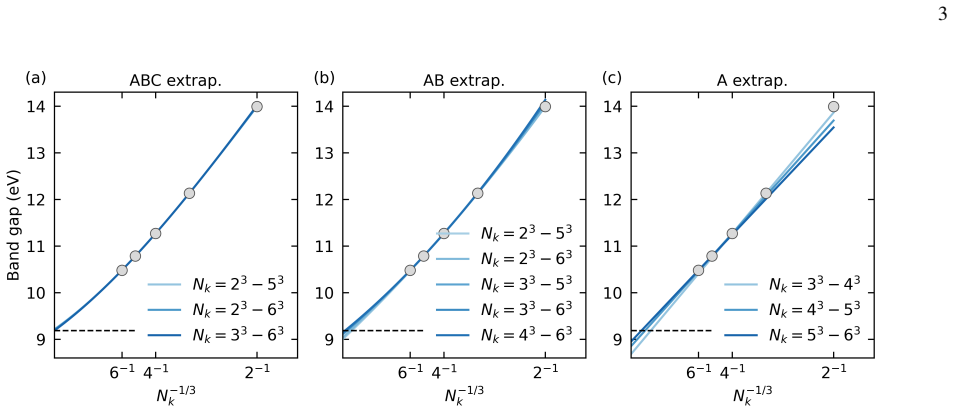
Direct band gaps We first evaluate the different extrapolation models, per- forming all calculations with PPs and the DZ basis. These results are summarized in Table III. When the ABC extrapola- tion model is used, band gaps obtained by including𝑁 𝑘 =2 3 as the smallest𝑘-mesh are generally close to those obtained from larger𝑘-mesh data sets. The largest d...
-
[3]
We consider the indirect band gap of BN as a representative example
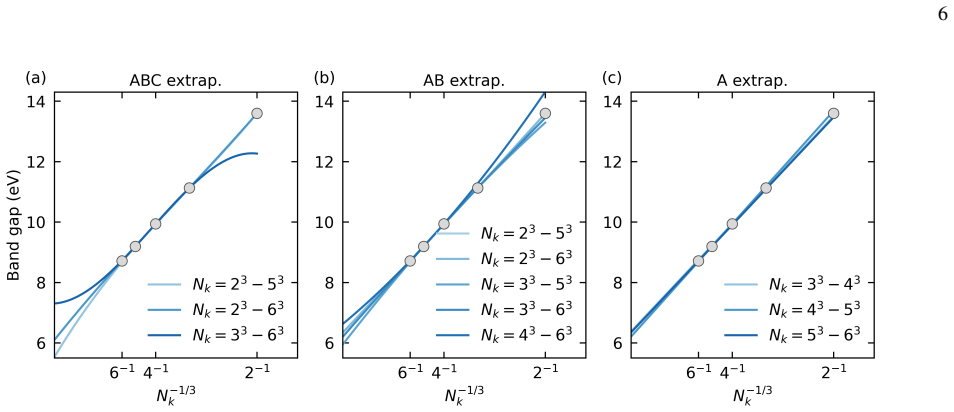
Indirect band gaps Next, we investigate the convergence behavior of indirect band gaps (using PPs and the DZ basis). We consider the indirect band gap of BN as a representative example. In BN, the VBM occurs at theΓpoint, and the CBM occurs as the X point. As shown in Fig. 3, over the𝑘-meshes accessible here, model A yields the most stable estimates, and ...
-
[4]
Final direct and indirect band gaps Using the same TDL extrapolation and basis-set correction procedures described above, we also perform AE-FC calcu- lations of direct and indirect band gaps. The only exception is LiF, for which the TZ and QZ calculations showed poor results using the settings described in Section II and conver- gence problems when tight...
-
[5]
Exploiting the space-group symmetry of the crystal reduces the number of independent𝑘-quartets from𝑁 3 𝑘 to𝑁 3 𝑘/G, whereGis the order of the point group
Space-group symmetry For a dense mesh with𝑁 𝑘 𝑘-points, two-electron quantities such as𝑡 𝑎𝑏 𝑖 𝑗 and the two-electron integrals are indexed by four 𝑘-points(k 𝑖,k 𝑗 ,k 𝑎,k 𝑏), of which only three are independent due to crystal momentum conservationk 𝑖 +k 𝑗 −k 𝑎 −k 𝑏 = G, whereGis a reciprocal lattice vector. Exploiting the space-group symmetry of the cryst...
-
[6]
Task scheduling and load balancing A naive distribution of IBZ quartets across processes leads to a severe load imbalance. The reason is that some BZ quartets coincide with stored IBZ blocks and can therefore be accessed with negligible overhead, while others must be reconstructed from their IBZ counterparts via the explicitO (𝑁 5)rotation described above...
-
[7]
Memory management Storing all two-electron quantities over the IBZ becomes prohibitively expensive for dense𝑘-point grids. To reduce memory usage, we store only the three-index density-fitting (DF) integrals𝐿 𝑄 𝑝𝑞 (replicated across processes) and compute all two-electron integral blocks involving at least two virtual orbitals (OVOV, VOOV, VOVV, and VVVV)...
-
[8]
All calcula- tions were performed on 96-core AMD Genoa nodes with 1.5 TB of memory per node
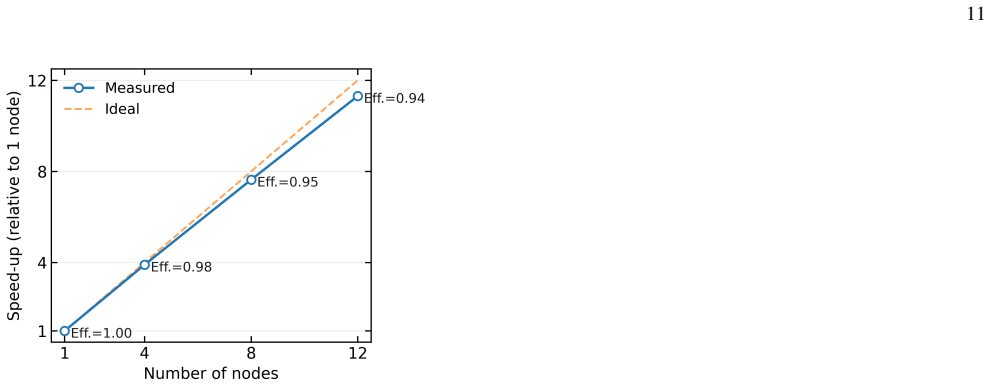
Performance We assess our parallel implementation using the ground- state CCSD calculation of rutile TiO2 on a 2×2×3𝑘-mesh, employing the GTH-DZVP-MOLOPT-SR basis. All calcula- tions were performed on 96-core AMD Genoa nodes with 1.5 TB of memory per node. We compute the average wall time per CCSD iteration with number of nodes from 1 to 12, and the speed...
-
[9]
Hirata, I
S. Hirata, I. Grabowski, M. Tobita, and R. J. Bartlett, Highly accurate treatment of electron correlation in polymers: Coupled- cluster and many-body perturbation theories, Chem. Phys. Lett. 345, 475 (2001)
2001
-
[10]
Hirata, R
S. Hirata, R. Podeszwa, M. Tobita, and R. J. Bartlett, Coupled- cluster singles and doubles for extended systems, J. Chem. Phys. 120, 2581 (2004)
2004
-
[11]
Katagiri, Equation-of-motion coupled-cluster study on exci- ton states of polyethylene with periodic boundary condition, J
H. Katagiri, Equation-of-motion coupled-cluster study on exci- ton states of polyethylene with periodic boundary condition, J. Chem. Phys.122, 224901 (2005)
2005
-
[12]
Gr¨ uneis, G
A. Gr¨ uneis, G. H. Booth, M. Marsman, J. Spencer, A. Alavi, and G. Kresse, Natural Orbitals for Wave Function Based Correlated Calculations Using a Plane Wave Basis Set, J. Chem. Theory Comput.7, 2780 (2011)
2011
-
[13]
McClain, Q
J. McClain, Q. Sun, G. K.-L. Chan, and T. C. Berkelbach, Gaussian-Based Coupled-Cluster Theory for the Ground-State and Band Structure of Solids, J. Chem. Theory Comput.13, 1209 (2017)
2017
-
[14]
S. J. Nolan, M. J. Gillan, D. Alf`e, N. L. Allan, and F. R. Manby, Calculation of properties of crystalline lithium hydride using correlated wave function theory, Phys. Rev. B80, 165109 (2009)
2009
-
[16]
Gruber, K
T. Gruber, K. Liao, T. Tsatsoulis, F. Hummel, and A. Gr¨ uneis, Applying the Coupled-Cluster Ansatz to Solids and Surfaces in the Thermodynamic Limit, Phys. Rev. X8, 021043 (2018)
2018
-
[17]
V. A. Neufeld, H.-Z. Ye, and T. C. Berkelbach, Ground-State Properties of Metallic Solids from Ab Initio Coupled-Cluster Theory, J. Phys. Chem. Lett.13, 7497 (2022)
2022
-
[18]
V. A. Neufeld and T. C. Berkelbach, Highly Accurate Electronic Structure of Metallic Solids from Coupled-Cluster Theory with Nonperturbative Triple Excitations, Phys. Rev. Lett.131, 186402 (2023)
2023
-
[19]
Ye and T
H.-Z. Ye and T. C. Berkelbach, Periodic Local Coupled-Cluster Theory for Insulators and Metals, J. Chem. Theory Comput.20, 8948 (2024)
2024
-
[20]
A. M. Lewis and T. C. Berkelbach,Ab InitioLinear and Pump– Probe Spectroscopy of Excitons in Molecular Crystals, J. Phys. Chem. Lett.11, 2241 (2020)
2020
-
[21]
Wang and T
X. Wang and T. C. Berkelbach, Excitons in Solids from Periodic Equation-of-Motion Coupled-Cluster Theory, J. Chem. Theory Comput.16, 3095 (2020)
2020
-
[22]
Y. Gao, Q. Sun, J. M. Yu, M. Motta, J. McClain, A. F. White, A. J. Minnich, and G. K.-L. Chan, Electronic structure of bulk manganese oxide and nickel oxide from coupled cluster theory, Phys. Rev. B101, 165138 (2020)
2020
-
[23]
Wang and T
X. Wang and T. C. Berkelbach, Absorption Spectra of Solids from Periodic Equation-of-Motion Coupled-Cluster Theory, J. Chem. Theory Comput.17, 6387 (2021)
2021
-
[24]
Gallo, F
A. Gallo, F. Hummel, A. Irmler, and A. Gr¨ uneis, A periodic equation-of-motion coupled-cluster implementation applied to F-centers in alkaline earth oxides, J. Chem. Phys.154, 064106 (2021)
2021
-
[25]
E. A. Vo, X. Wang, and T. C. Berkelbach, Performance of pe- riodic EOM-CCSD for bandgaps of inorganic semiconductors and insulators, J. Chem. Phys.160, 044106 (2024)
2024
-
[26]
Moerman, A
E. Moerman, A. Gallo, A. Irmler, T. Sch ¨afer, F. Hummel, A. Gr¨ uneis, and M. Scheffler, Finite-Size Effects in Periodic EOM-CCSD for Ionization Energies and Electron Affinities: Convergence Rate and Extrapolation to the Thermodynamic Limit, J. Chem. Theory Comput.21, 1865 (2025)
2025
-
[27]
Moerman, H
E. Moerman, H. Miranda, A. Gallo, A. Irmler, T. Sch ¨afer, F. Hummel, M. Engel, G. Kresse, M. Scheffler, and A. Gr¨ uneis, Exploring the accuracy of the equation-of-motion coupled- cluster band gap of solids, Phys. Rev. B111, L121202 (2025)
2025
-
[28]
E. A. Vo and T. C. Berkelbach, Core binding energies of solids with periodic EOM-CCSD (2025), arXiv:2508.00168 [physics]
arXiv 2025
-
[29]
T. N. Mihm, L. Weiler, and J. J. Shepherd, How the Exchange Energy Can Affect the Power Laws Used to Extrapolate the Cou- pled Cluster Correlation Energy to the Thermodynamic Limit, J. Chem. Theory Comput.19, 1686 (2023)
2023
-
[30]
Xing and L
X. Xing and L. Lin, Inverse Volume Scaling of Finite-Size Error in Periodic Coupled Cluster Theory, Phys. Rev. X14, 011059 (2024)
2024
-
[31]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: The Python-based sim- ulations of chemistry framework, WIREs Comput. Mol. Sci.8, e1340 (2018)
2018
-
[32]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y. Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Ko- val, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. Sc...
2020
-
[33]
Q. Sun, M. R. Hermes, X. Wu, H. Zhai, X. Zhang, A. M. Ahmed, J. J. Aucar, O. J. Backhouse, S. Banerjee, P. Bao, N. A. Bogdanov, K. Bystrom, F. Chapoton, N.-Y. Chen, I. Y. Chernyshov, H. S. Clifford, S. Cohen-Janes, Z.-H. Cui, Y. D. Damour, N. Dattani, L. B. Dittmer, S. Ehlert, J. J. Eriksen, F. A. Evangelista, S. A. Ewing, A. Farahvash, K. Focke, Y. Gao, ...
Pith/arXiv arXiv 2026
-
[34]
Q. Sun, T. C. Berkelbach, J. D. McClain, and G. K.-L. Chan, Gaussian and plane-wave mixed density fitting for periodic sys- tems, J. Chem. Phys.147, 164119 (2017)
2017
-
[35]
Ye and T
H.-Z. Ye and T. C. Berkelbach, Fast periodic Gaussian density fitting by range separation, J. Chem. Phys.154, 131104 (2021)
2021
-
[36]
Paier, R
J. Paier, R. Hirschl, M. Marsman, and G. Kresse, The Perdew– Burke–Ernzerhof exchange-correlation functional applied to the 13 G2-1 test set using a plane-wave basis set, J. Chem. Phys.122, 234102 (2005)
2005
-
[37]
Broqvist, A
P. Broqvist, A. Alkauskas, and A. Pasquarello, Hybrid- functional calculations with plane-wave basis sets: Effect of singularity correction on total energies, energy eigenvalues, and defect energy levels, Phys. Rev. B80, 085114 (2009)
2009
-
[38]
Sundararaman and T
R. Sundararaman and T. A. Arias, Regularization of the Coulomb singularity in exact exchange by Wigner-Seitz trun- cated interactions: Towards chemical accuracy in nontrivial systems, Phys. Rev. B87, 165122 (2013)
2013
-
[39]
T. H. Dunning, Gaussian basis sets for use in correlated molecu- lar calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys.90, 1007 (1989)
1989
-
[40]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B54, 1703 (1996)
1996
-
[41]
Hartwigsen, S
C. Hartwigsen, S. Goedecker, and J. Hutter, Relativistic separa- ble dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B58, 3641 (1998)
1998
-
[42]
Ye and T
H.-Z. Ye and T. C. Berkelbach, Correlation-Consistent Gaussian Basis Sets for Solids Made Simple, J. Chem. Theory Comput. 18, 1595 (2022)
2022
-
[43]
C. Sosa, J. Geertsen, G. W. Trucks, R. J. Bartlett, and J. A. Franz, Selection of the reduced virtual space for correlated calculations. An application to the energy and dipole moment of H2O, Chem. Phys. Lett.159, 148 (1989)
1989
-
[44]
A. G. Taube and R. J. Bartlett, Frozen Natural Orbitals: System- atic Basis Set Truncation for Coupled-Cluster Theory, Collect. Czech. Chem. Commun70, 837 (2005)
2005
-
[45]
Landau, K
A. Landau, K. Khistyaev, S. Dolgikh, and A. I. Krylov, Frozen natural orbitals for ionized states within equation-of-motion coupled-cluster formalism, J. Chem. Phys.132, 014109 (2010)
2010
-
[46]
A. E. I. DePrince and C. D. Sherrill, Accurate Noncovalent In- teraction Energies Using Truncated Basis Sets Based on Frozen Natural Orbitals, J. Chem. Theory Comput.9, 293 (2013)
2013
-
[47]
A. E. I. DePrince and C. D. Sherrill, Accuracy and Efficiency of Coupled-Cluster Theory Using Density Fitting/Cholesky De- composition, Frozen Natural Orbitals, and a t1-Transformed Hamiltonian, J. Chem. Theory Comput.9, 2687 (2013)
2013
-
[48]
Goldzak, X
T. Goldzak, X. Wang, H.-Z. Ye, and T. C. Berkelbach, Accu- rate thermochemistry of covalent and ionic solids from spin- component-scaled MP2, J. Chem. Phys.157, 174112 (2022)
2022
-
[49]
Kresse and J
G. Kresse and J. Hafner, Norm-conserving and ultrasoft pseu- dopotentials for first-row and transition elements, J. Phys.: Con- dens. Matter6, 8245 (1994)
1994
-
[50]
Kresse and J
G. Kresse and J. Furthm¨ uller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B54, 11169 (1996)
1996
-
[51]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler,Ab Initiomolecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun. 180, 2175 (2009)
2009
-
[52]
Moerman, F
E. Moerman, F. Hummel, A. Gr¨ uneis, A. Irmler, and M. Schef- fler, Interface to high-performance periodic coupled-cluster the- ory calculations with atom-centered, localized basis functions, J. Open Source Softw.7, 4040 (2022)
2022
-
[53]
Zhu and G
T. Zhu and G. K.-L. Chan, All-Electron Gaussian-BasedG 0 W0 for Valence and Core Excitation Energies of Periodic Systems, J. Chem. Theory Comput.17, 727 (2021)
2021
-
[54]
M. S. Hybertsen and S. G. Louie,Ab Initiostatic dielectric matrices from the density-functional approach. I. Formulation and application to semiconductors and insulators, Phys. Rev. B 35, 5585 (1987)
1987
-
[55]
Zhang, A
G.-X. Zhang, A. M. Reilly, A. Tkatchenko, and M. Scheffler, Performance of various density-functional approximations for cohesive properties of 64 bulk solids, New J. Phys.20, 063020 (2018)
2018
-
[56]
Schimka, J
L. Schimka, J. Harl, and G. Kresse, Improved hybrid functional for solids: The HSEsol functional, J. Chem. Phys.134, 024116 (2011)
2011
-
[57]
J. Zhang, M.-F. Chen, A. Rettig, T. Jiang, P. J. Robinson, H. Q. Dinh, A. Z. Ni, and J. Lee, Ab Initio Auxiliary-Field Quantum Monte Carlo in the Thermodynamic Limit (2026), arXiv:2602.16679 [cond-mat]
arXiv 2026
-
[58]
F. Tran, J. Stelzl, and P. Blaha, Rungs 1 to 4 of DFT Jacob’s ladder: Extensive test on the lattice constant, bulk modulus, and cohesive energy of solids, J. Chem. Phys.144, 204120 (2016)
2016
-
[59]
Engel, H
M. Engel, H. Miranda, L. Chaput, A. Togo, C. Verdi, M. Mars- man, and G. Kresse, Zero-point renormalization of the band gap of semiconductors and insulators using the projector augmented wave method, Phys. Rev. B106, 094316 (2022)
2022
-
[60]
Chiang,Electronic structure of solids: Photoemission spectra and related data(Springer, 1989)
T. Chiang,Electronic structure of solids: Photoemission spectra and related data(Springer, 1989)
1989
-
[61]
Madelung,Semiconductors: Data Handbook, Vol
O. Madelung,Semiconductors: Data Handbook, Vol. 385 (Springer, 2004)
2004
-
[62]
M. E. Levinshtein, S. L. Rumyantsev, and M. S. Shur,Properties of Advanced Semiconductor Materials: GaN, AIN, InN, BN, SiC, SiGe(John Wiley & Sons, 2001)
2001
-
[63]
K. Woo, K. Lee, and K. Kovnir, BP: Synthesis and properties of boron phosphide, Mater. Res. Express3, 074003 (2016)
2016
-
[64]
Baroni, G
S. Baroni, G. Pastori Parravicini, and G. Pezzica, Quasiparticle band structure of lithium hydride, Phys. Rev. B32, 4077 (1985)
1985
-
[65]
Piacentini, D
M. Piacentini, D. W. Lynch, and C. G. Olson, Thermoreflectance of LiF between 12 and 30 eV, Phys. Rev. B13, 5530 (1976)
1976
-
[66]
Baldini and B
G. Baldini and B. Bosacchi, Optical Properties of Na and Li Halide Crystals at 55◦K, physica status solidi (b)38, 325 (1970)
1970
-
[67]
R. C. Whited, C. J. Flaten, and W. C. Walker, Exciton ther- moreflectance of MgO and CaO, Solid State Commun.13, 1903 (1973)
1903
-
[68]
Schneider, M
J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo, and D. W. Bahnemann, Understanding TiO 2 Photo- catalysis: Mechanisms and Materials, Chem. Rev.114, 9919 (2014)
2014
-
[69]
Wang and K
Q. Wang and K. Domen, Particulate Photocatalysts for Light- Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev.120, 919 (2020)
2020
-
[70]
D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, Michael. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh, and A. A. Sokol, Band alignment of rutile and anatase TiO2, Nat. Mater.12, 798 (2013)
2013
-
[71]
Rangan, S
S. Rangan, S. Katalinic, R. Thorpe, R. A. Bartynski, J. Rochford, and E. Galoppini, Energy Level Alignment of a Zinc(II) Tetraphenylporphyrin Dye Adsorbed onto TiO2(110) and ZnO(1120) Surfaces, J. Phys. Chem. C114, 1139 (2010)
2010
-
[72]
Y.-N. Wu, W. A. Saidi, P. Ohodnicki, B. Chorpening, and Y. Duan, First-Principles Investigations of the Temperature De- pendence of Electronic Structure and Optical Properties of Ru- tile TiO2, J. Phys. Chem. C122, 22642 (2018)
2018
-
[73]
M. E. Arroyo-de Dompablo, A. Morales-Garc ´ıa, and M. Tar- avillo, DFT+Ucalculations of crystal lattice, electronic struc- ture, and phase stability under pressure of TiO2 polymorphs, J. Chem. Phys.135, 054503 (2011)
2011
-
[74]
Kang and M
W. Kang and M. S. Hybertsen, Quasiparticle and optical prop- erties of rutile and anatase TiO 2, Phys. Rev. B82, 085203 (2010)
2010
-
[75]
Landmann, E
M. Landmann, E. Rauls, and W. G. Schmidt, The electronic structure and optical response of rutile, anatase and brookite 14 TiO2, J. Phys.: Condens. Matter24, 195503 (2012)
2012
-
[76]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, and K. A. Persson, Commentary: The Materials Project: A materi- als genome approach to accelerating materials innovation, APL Mater.1, 011002 (2013)
2013
-
[77]
Gr¨ uneis, M
A. Gr¨ uneis, M. Marsman, and G. Kresse, Second-order Møller– Plesset perturbation theory applied to extended systems. II. Structural and energetic properties, J. Chem. Phys.133, 074107 (2010)
2010
-
[78]
I. Y. Zhang, A. J. Logsdail, X. Ren, S. V. Levchenko, L. Ghir- inghelli, and M. Scheffler, Main-group test set for materials science and engineering with user-friendly graphical tools for error analysis: Systematic benchmark of the numerical and in- trinsic errors in state-of-the-art electronic-structure approxima- tions, New J. Phys.21, 013025 (2019)
2019
-
[79]
Reaching the thermodynamic limit of periodic CCSD cohesive energies and band gaps with denser Brillouin zone sampling
M. Nooijen and R. J. Bartlett, Equation of motion coupled clus- ter method for electron attachment, J. Chem. Phys.102, 3629 (1995). Supplemental Material for “Reaching the thermodynamic limit of periodic CCSD cohesive energies and band gaps with denser Brillouin zone sampling” Shuhang Li,1 Huanchen Zhai,2 Francesco A. Evangelista, 1 and Timothy C. Berkelb...
1995
-
[80]
Gr¨ uneis, M
A. Gr¨ uneis, M. Marsman, and G. Kresse, Second-order Møller–Plesset perturbation theory applied to extended systems. II. Structural and energetic properties, J. Chem. Phys.133, 074107 (2010)
2010
-
[81]
G. H. Booth, A. Gr¨ uneis, G. Kresse, and A. Alavi, Towards an exact description of electronic wavefunctions in real solids, Nature493, 365 (2013)
2013
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.