Reduced-Order Modelling of Defect Transport using Surrogate Kinetics: Application to U_xPu_ {1-x}N
Pith reviewed 2026-06-26 07:39 UTC · model grok-4.3
The pith
Surrogate functions from local coordination counts accurately predict migration barriers for defects in UxPu1-xN.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
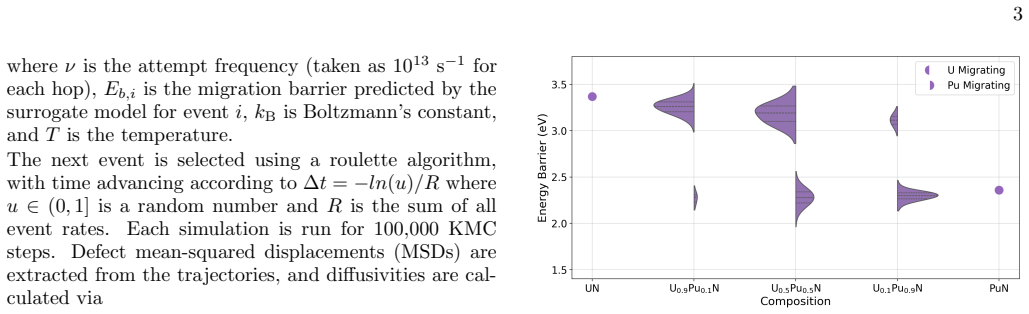
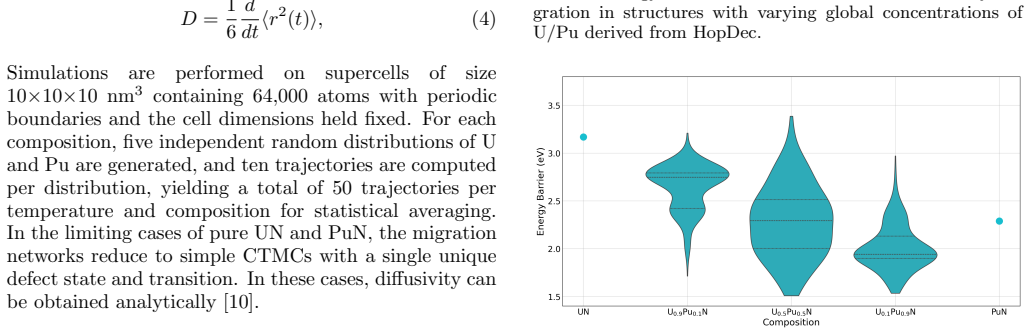
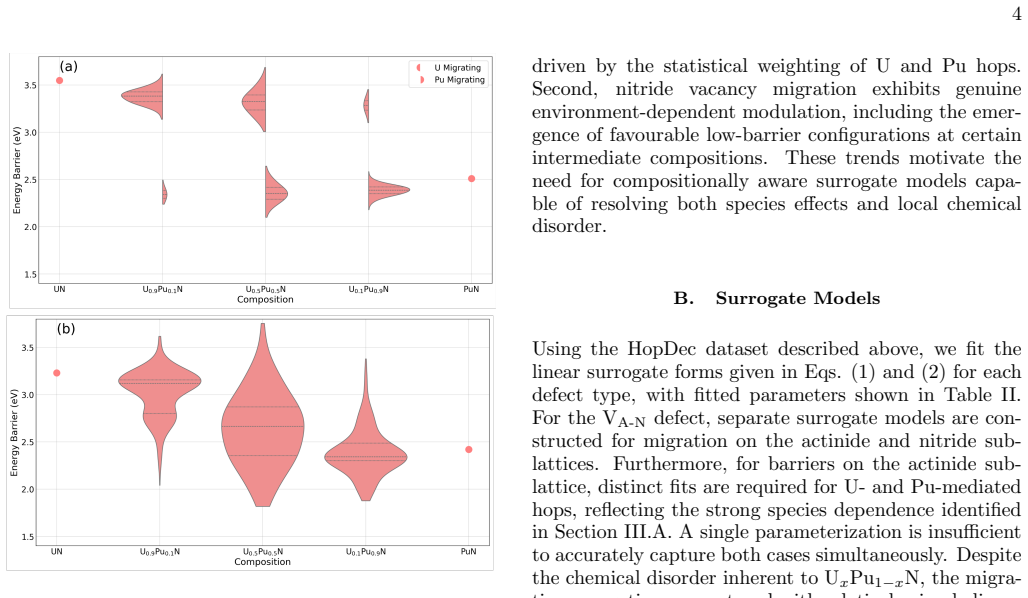
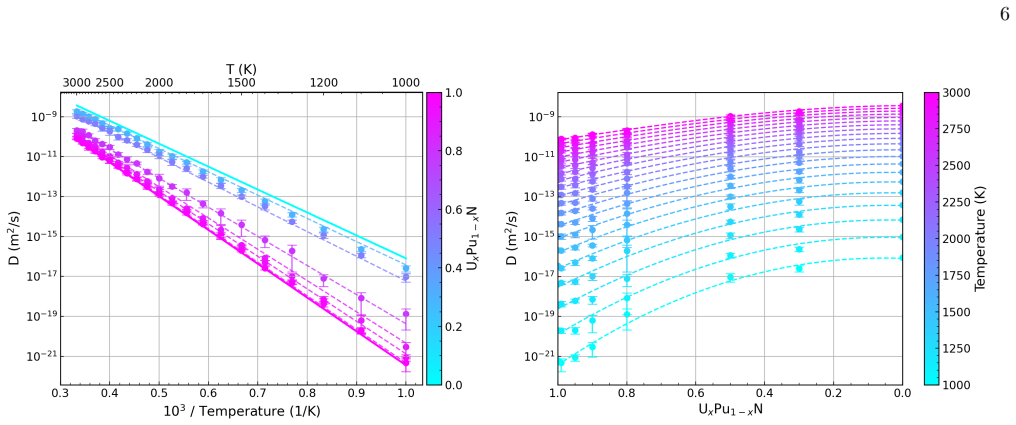
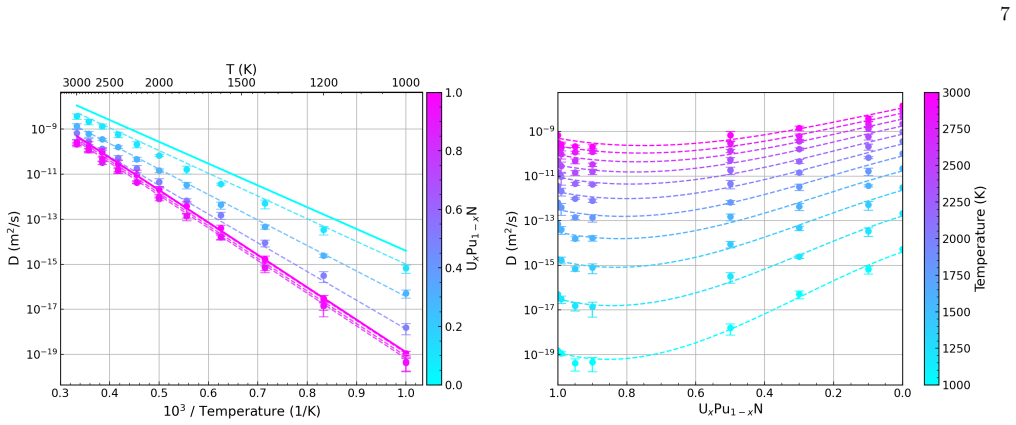
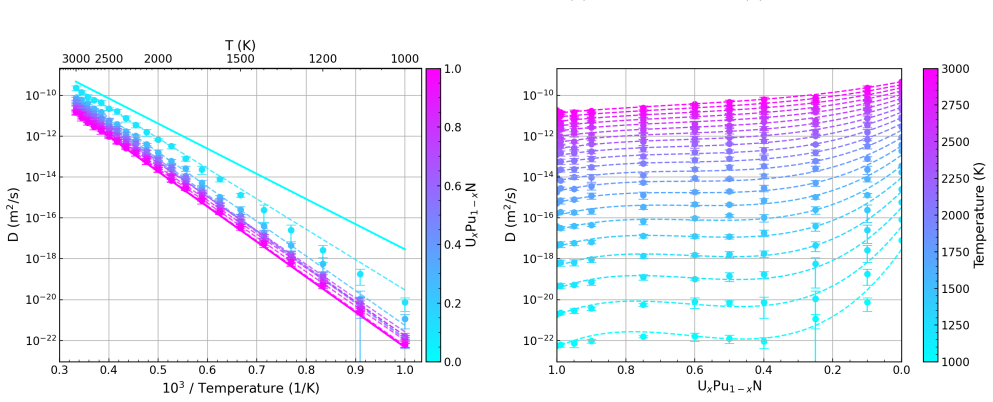
Migration energetics for actinide vacancies, nitride vacancies, and actinide-nitride divacancies in UxPu1-xN are generated across thousands of chemical configurations and then distilled into compact surrogate functions based on local coordination counts. The surrogates reproduce the atomistic dataset with low error and enable efficient evaluation of migration rates during lattice kinetic Monte Carlo simulations. Long-time diffusivities computed across temperature and composition reveal strongly non-linear behaviour arising from species-controlled migration on the actinide sublattice and environment-dependent trapping on the nitride sublattice.
What carries the argument
Environment-dependent surrogate kinetics that map local coordination counts to migration barriers and energy differences.
If this is right
- Surrogate models reproduce the atomistic dataset with low error and support efficient migration-rate evaluation inside lattice kinetic Monte Carlo.
- Long-time diffusivities can be obtained across ranges of temperature and composition without enumerating every possible local environment.
- Diffusion exhibits strongly non-linear dependence on temperature and composition due to two dominant mechanisms.
- The same reduced-order approach supplies a general route to defect-transport modelling in other chemically disordered materials.
Where Pith is reading between the lines
- The surrogate approach could be inserted into continuum-scale fuel-performance codes to carry atomistic kinetics upward without prohibitive cost.
- Experimental measurements of composition-dependent diffusivities in UxPu1-xN would provide an external check on the two identified mechanisms.
- If local-count descriptors prove insufficient for a new material, the same workflow could test whether adding one or two extra descriptors restores accuracy.
Load-bearing premise
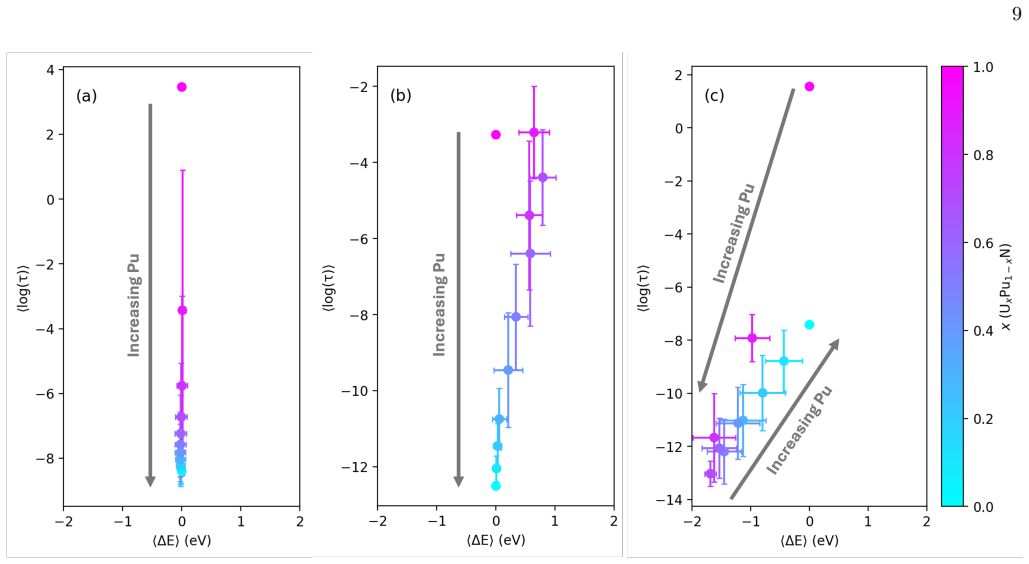
Descriptors based solely on local coordination counts are sufficient to capture the full distribution of transition barriers and energy differences without loss of essential physics.
What would settle it
Direct comparison of surrogate-driven kinetic Monte Carlo diffusivities against independent atomistic calculations performed on a composition or temperature range not used to train the surrogates.
Figures

read the original abstract
Defect transport in chemically disordered materials is a difficult phenomenon to model since migration energetics depend strongly on the local chemical environment, producing a distribution of transition barriers that cannot be exhaustively enumerated. Here, we develop a reduced-order approach to modelling defect diffusion in compositionally complex materials using environment-dependent surrogate kinetics. Migration energetics for actinide vacancies, nitride vacancies, and actinide-nitride divacancies in U$_x$Pu$_{1-x}$N are generated using the Hop-Decorate workflow across thousands of chemical configurations. These data are distilled into compact surrogate functions that predict migration barriers and energy differences from simple descriptors based on local coordination counts. The surrogate models reproduce the atomistic dataset with low error and enable efficient evaluation of migration rates during lattice kinetic Monte Carlo simulations. Long-time diffusivities computed across temperature and composition reveal strongly non-linear behaviour arising from two dominant mechanisms: species-controlled migration on the actinide sublattice and environment-dependent trapping on the nitride sublattice. Although demonstrated for U$_x$Pu$_{1-x}$N the framework provides a general and computationally efficient approach for modelling defect transport in chemically disordered materials and for integrating atomistic kinetics into higher-scale simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a reduced-order modeling framework for defect transport in chemically disordered UxPu1-xN using environment-dependent surrogate kinetics. Atomistic migration energetics for actinide vacancies, nitride vacancies, and divacancies are generated via the Hop-Decorate workflow across thousands of configurations and distilled into compact surrogate functions that predict barriers and energy differences from local coordination count descriptors. These surrogates are inserted into lattice kinetic Monte Carlo simulations to compute long-time diffusivities, which exhibit strongly non-linear dependence on temperature and composition arising from species-controlled actinide-sublattice migration and environment-dependent nitride-sublattice trapping. The approach is positioned as a general, efficient method for integrating atomistic kinetics into higher-scale models of disordered materials.
Significance. If the central claims hold, the work supplies a practical route to embed atomistic-scale chemical-environment effects into mesoscale transport simulations without exhaustive enumeration of barriers. The demonstration of non-linear diffusivity trends underscores the necessity of local-environment modeling in mixed actinide nitrides. Credit is due for the systematic generation of a large atomistic dataset via Hop-Decorate and for the explicit construction of surrogate functions that enable direct KMC evaluation.
major comments (2)
- [Abstract] Abstract: the claim that 'the surrogate models reproduce the atomistic dataset with low error' is unsupported by any quantitative metric (MAE, RMSE, R², or error bars), cross-validation procedure, or test-set performance; without these numbers the assertion that the surrogates are adequate for KMC migration-rate evaluation remains unverified and is load-bearing for the reported non-linear diffusivity results.
- [Abstract] Abstract (and implied methods): the central modeling assumption that descriptors based solely on local coordination counts capture the full distribution of transition barriers and energy differences is not shown to be sufficient. Longer-range elastic or electronic interactions that vary with U/Pu mixing in the rocksalt lattice could produce systematic outliers or shifts; if present, these would bias the KMC rates and undermine the claimed non-linear trends even if average training-set error appears acceptable.
minor comments (2)
- Notation for the surrogate functions and the precise definition of the coordination-count descriptors should be introduced with explicit equations rather than left implicit.
- The temperature and composition ranges over which the non-linear diffusivity behavior is observed should be stated quantitatively in the abstract or early results section.
Simulated Author's Rebuttal
We thank the referee for their thorough review and constructive comments on our manuscript. We address each major comment below, indicating revisions where the manuscript will be strengthened.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that 'the surrogate models reproduce the atomistic dataset with low error' is unsupported by any quantitative metric (MAE, RMSE, R², or error bars), cross-validation procedure, or test-set performance; without these numbers the assertion that the surrogates are adequate for KMC migration-rate evaluation remains unverified and is load-bearing for the reported non-linear diffusivity results.
Authors: We agree that the abstract statement would be strengthened by explicit quantitative support. The full manuscript reports MAE, RMSE, and R² values together with cross-validation and test-set results in the surrogate-model construction section; these metrics confirm low error and support KMC use. We will revise the abstract to include the key performance numbers (MAE and R²) and a brief reference to the validation procedure. revision: yes
-
Referee: [Abstract] Abstract (and implied methods): the central modeling assumption that descriptors based solely on local coordination counts capture the full distribution of transition barriers and energy differences is not shown to be sufficient. Longer-range elastic or electronic interactions that vary with U/Pu mixing in the rocksalt lattice could produce systematic outliers or shifts; if present, these would bias the KMC rates and undermine the claimed non-linear trends even if average training-set error appears acceptable.
Authors: The referee correctly identifies a modeling assumption whose sufficiency is not exhaustively proven. Our Hop-Decorate sampling across thousands of configurations shows that local coordination counts explain the dominant variance in barriers for this rocksalt system, yielding the reported non-linear diffusivity trends. Nevertheless, we will add a concise limitations paragraph in the Discussion that acknowledges possible longer-range contributions and states that the current descriptor set is a pragmatic, computationally efficient choice validated on the generated dataset. No new calculations are required for this clarification. revision: partial
Circularity Check
No significant circularity in derivation chain
full rationale
The paper generates atomistic migration energetics via the Hop-Decorate workflow across chemical configurations, fits surrogate functions on local coordination descriptors to reproduce that dataset, and then applies the surrogates in independent lattice KMC runs to compute long-time diffusivities across temperature and composition. The diffusivities are not inputs to the surrogate fitting, and no equations or steps reduce the final results to the training data by construction. No self-citations, uniqueness theorems, or ansatzes imported from prior author work are referenced in the text. The derivation chain remains self-contained against external atomistic benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Local coordination counts suffice as descriptors for migration barriers and energy differences
Reference graph
Works this paper leans on
-
[1]
Actinide vacancy diffusion is dominated by species- controlled kinetics (U vs Pu hops), producing strong non-linearity and a rapid rise in mobility once sufficiently many fast events contribute to long-range motion
-
[2]
Nitride vacancy diffusion is governed by environment-sensitive disorder effects, with evidence for both trap-dominated regimes at low Pu additions, known as sluggish diffusion, and enhanced mobility at higher Pu where lower barriers become more prevalent and/or better connected, namely percolation
-
[3]
Divacancy diffusion reflects coupled sampling of both sublattices, showing behaviour that can be qualitatively distinct from isolated vacancy trans- port and emphasizing the importance of mechanism competition within the bound complex. Fits to these data derive the following equations for dif- fusivity as a function of temperature and concentration, 8 x: ...
-
[4]
This is likely due to the complex heterogenous potential energy landscapes created by the compositional variations, however, a few interesting findings arise from analysis
Much of the data has large error bars and the ma- jority of the time a consistent trend cannot be reliably identified. This is likely due to the complex heterogenous potential energy landscapes created by the compositional variations, however, a few interesting findings arise from analysis. For the VA defect, there is a simple relationship in which, with ...
-
[5]
Nastar and F
M. Nastar and F. Soisson, Comprehensive nuclear mate- rials1, 471 (2012)
2012
-
[6]
Nichols, Journal of Nuclear Materials69, 451 (1978)
F. Nichols, Journal of Nuclear Materials69, 451 (1978)
1978
-
[7]
Rajasekaran, P
G. Rajasekaran, P. Narayanan, and A. Parashar, Crit- ical reviews in solid state and materials sciences41, 47 (2016)
2016
-
[8]
Xiao and I
H. Xiao and I. Baker, Acta metallurgica et materialia43, 391 (1995)
1995
-
[9]
Faulkner and C
D. Faulkner and C. Woo, Journal of Nuclear Materials 90, 307 (1980)
1980
-
[10]
A. F. Voter, inRadiation effects in solids(Springer, 2007) pp. 1–23
2007
-
[11]
C. L. Bris, T. Lelievre, M. Luskin, and D. Perez, Monte Carlo Methods Appl.18, 119 (2012)
2012
-
[12]
Novotny, Physical review letters74, 1 (1995)
M. Novotny, Physical review letters74, 1 (1995)
1995
-
[13]
Van der Ven, J
A. Van der Ven, J. C. Thomas, B. Puchala, and A. R. Natarajan, Annual Review of Materials Research48, 27 (2018)
2018
-
[14]
T. D. Swinburne and D. Perez, NPJ Computational Ma- terials6, 190 (2020)
2020
-
[15]
Ebmeyer and P
W. Ebmeyer and P. P. Dholabhai, Materials Advances (2024)
2024
-
[16]
Becquart and C
C. Becquart and C. Domain, Journal of Nuclear Materi- als385, 223 (2009)
2009
-
[17]
Perez, E
D. Perez, E. D. Cubuk, A. Waterland, E. Kaxiras, and A. F. Voter, Journal of chemical theory and computation 12, 18 (2016)
2016
-
[18]
K¨ orfer, A
S. K¨ orfer, A. Bonkowski, J. Kler, P. Hatton, B. P. Uberu- aga, and R. A. De Souza, Advanced Engineering Mate- rials25, 2201788 (2023)
2023
-
[19]
Vaari, Solid State Ionics270, 10 (2015)
J. Vaari, Solid State Ionics270, 10 (2015)
2015
-
[20]
Schneider, C
A. Schneider, C. Matthews, D. Andersson, and M. Cooper, Journal of Nuclear Materials , 156360 (2025)
2025
-
[21]
A. A. Talapatra, A. Pandey, M. S. Wilson, Y. W. Li, G. Pilania, B. P. Uberuaga, and D. Perez, Computer Physics Communications , 109752 (2025)
2025
-
[22]
Huang and X.-M
W. Huang and X.-M. Bai, Materialia , 102531 (2025)
2025
-
[23]
B. Xu, J. Zhang, S. Ma, Y. Xiong, S. Huang, J. Kai, and S. Zhao, Acta Materialia234, 118051 (2022)
2022
-
[24]
S. Zhao, Y. Zhang, and W. J. Weber, Journal of Nuclear Materials561, 153573 (2022)
2022
-
[25]
B. Xing, B. Xie, W. Zou, E. Lang, E. Boltynjuk, H. Chen, M. P. Short, G. Tynan, T. J. Rupert, J. Trelewicz,et al., arXiv preprint arXiv:2603.02651 (2026)
arXiv 2026
-
[26]
Hatton and B
P. Hatton and B. P. Uberuaga, Journal of Materials Chemistry A (2023)
2023
-
[27]
Ekberg, D
C. Ekberg, D. Ribeiro Costa, M. Hedberg, and M. Jolkkonen, Journal of radioanalytical and nuclear chemistry318, 1713 (2018)
2018
-
[28]
M. Uno, T. Nishi, and M. Takano, inComprehensive 13 Nuclear Materials: Second Edition(2020) pp. 202–231
2020
-
[29]
AbdulHameed, B
M. AbdulHameed, B. Beeler, C. O. Galvin, M. W. Cooper, N. Elamrawy, and A. Claisse, Journal of Nu- clear Materials , 156153 (2025)
2025
-
[30]
R. D. Syarifah and Z. Suud, inAIP Conference Proceed- ings, Vol. 1677 (AIP Publishing LLC, 2015) p. 120005
2015
-
[31]
Y. Arai, M. Morihira, and T. Ohmichi, Journal of nuclear materials202, 70 (1993)
1993
-
[32]
Zullo, D
G. Zullo, D. Pizzocri, and L. Luzzi, Journal of Nuclear Materials587, 154744 (2023)
2023
-
[33]
J. Peng, W. R. Deskins, L. Malakkal, and A. El-Azab, Journal of Applied Physics130(2021)
2021
-
[34]
Galvin, N
C. Galvin, N. Kuganathan, N. J. Barron, and R. W. Grimes, Journal of Applied Physics135(2024)
2024
-
[35]
Hatton, B
P. Hatton, B. P. Uberuaga, and D. Perez, Computational Materials Science259, 114165 (2025)
2025
-
[36]
Hop-Decorate,
P. Hatton, “Hop-Decorate,”https://github.com/ PeteHatton/Hop-Decorate(2024)
2024
-
[37]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen,et al., Com- puter Physics Communications271, 108171 (2022)
2022
-
[38]
Hjorth Larsen, J
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus,et al., Journal of Physics: Condensed Matter29, 273002 (2017)
2017
-
[39]
Kocevski, M
V. Kocevski, M. W. Cooper, A. J. Claisse, and D. A. Andersson, Journal of Nuclear Materials562, 153553 (2022)
2022
-
[40]
G. Henkelman, B. P. Uberuaga, and H. J´ onsson, The Journal of Chemical Physics113, 9901 (2000), https://doi.org/10.1063/1.1329672
-
[41]
J. J. Li, N. Zagni, W. D. Neilson, R. L. Gray, and S. T. Murphy, Journal of Nuclear Materials586, 154656 (2023)
2023
-
[42]
Y. N. Osetsky, L. K. B´ eland, A. V. Barashev, and Y. Zhang, Current Opinion in Solid State and Materi- als Science22, 65 (2018)
2018
-
[43]
Tsai, M.-H
K.-Y. Tsai, M.-H. Tsai, and J.-W. Yeh, Acta Materialia 61, 4887 (2013)
2013
-
[44]
Dkabrowa, W
J. Dkabrowa, W. Kucza, G. Cie´ slak, T. Kulik, M. Danielewski, and J.-W. Yeh, Journal of Alloys and Compounds674, 455 (2016)
2016
-
[45]
Matthews, R
C. Matthews, R. Perriot, M. W. Cooper, C. R. Stanek, and D. A. Andersson, Journal of Nuclear Materials527, 151787 (2019)
2019
-
[46]
Moggridge, Chemical engineering science71, 226 (2012)
G. Moggridge, Chemical engineering science71, 226 (2012)
2012
-
[47]
Adjanor, M
G. Adjanor, M. Ath` enes, C. Domain, and N. Mousseau, inEPJ Web of Conferences, Vol. 302 (EDP Sciences,
-
[48]
Hatton, P
P. Hatton, P. Goddard, R. Smith, A. Abbas, C. Potami- alis, R. Greenhalgh, and J. Walls, Thin Solid Films692, 137614 (2019)
2019
-
[49]
D. R. Mason, A. E. Sand, and S. L. Dudarev, Modelling and simulation in materials science and engineering27, 055003 (2019)
2019
-
[50]
B. P. Uberuaga, E. Mart´ ınez, D. Perez, and A. F. Voter, Computational Materials Science147, 282 (2018)
2018
-
[51]
Vincent, C
E. Vincent, C. Becquart, and C. Domain, Journal of nuclear materials359, 227 (2006)
2006
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.