PolyGraphPy: A unified Python framework for atomistic simulation and machine learning-driven polymer design
Pith reviewed 2026-06-28 00:32 UTC · model grok-4.3
The pith
PolyGraphPy integrates DFTB simulations with Bayesian GNNs and generative models for polymer property prediction and design.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
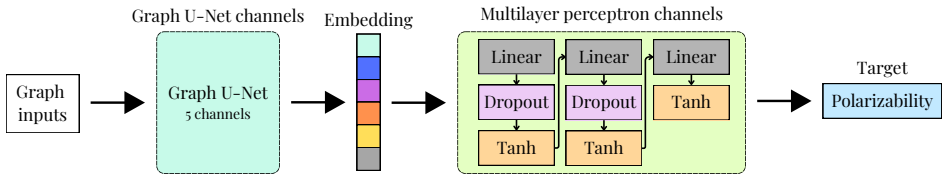
PolyGraphPy supplies a unified platform that links efficient atomistic simulations for dataset generation with Bayesian GNN property predictors and complementary generative models for targeted polymer creation, shown to work on acrylates.
What carries the argument
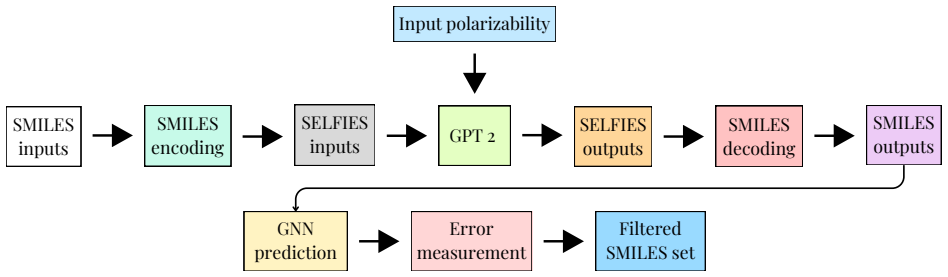
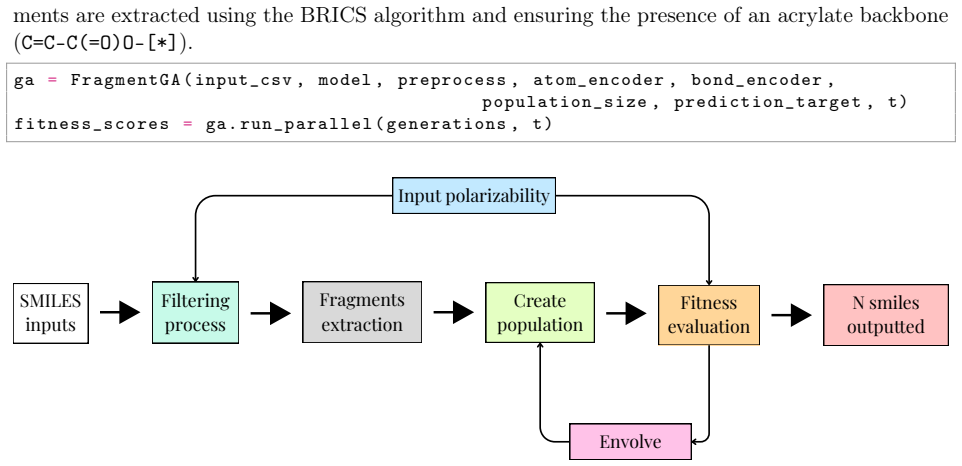
Bayesian Graph Neural Networks using stochastic graph representations for property prediction and uncertainty quantification, paired with SELFIES-GPT and BRICS-GA generative models.
Load-bearing premise
Bayesian Graph Neural Networks with stochastic graph representations deliver accurate predictions and robust uncertainty quantification for properties such as static polarizability.
What would settle it
Comparison of Bayesian GNN predictions for static polarizability against experimental measurements on a held-out set of acrylate polymers.
Figures

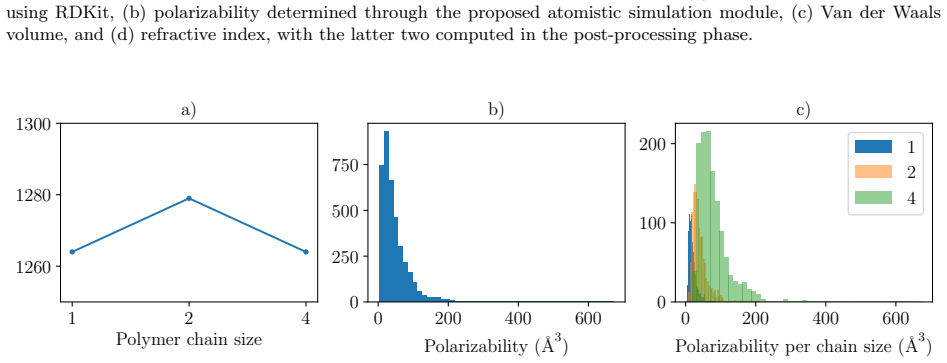
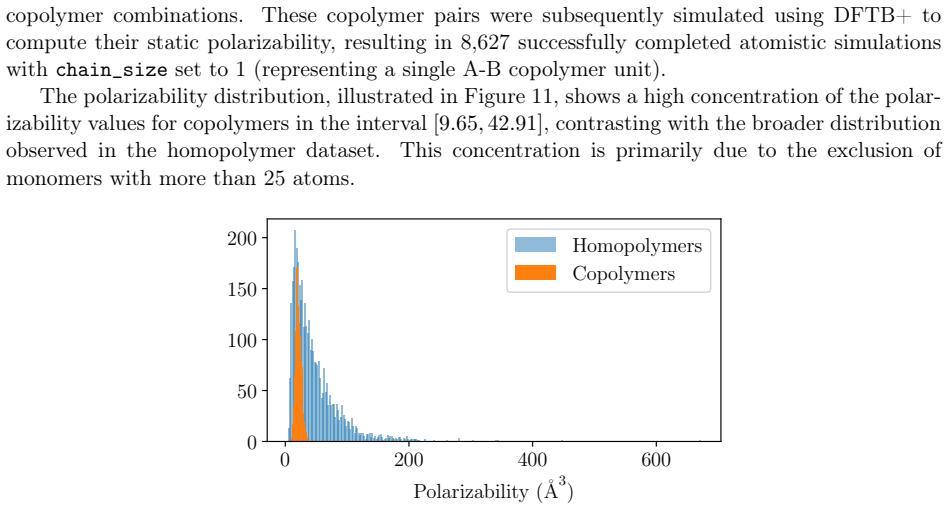
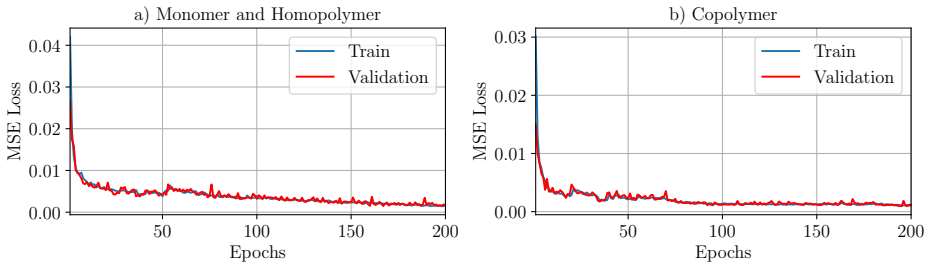
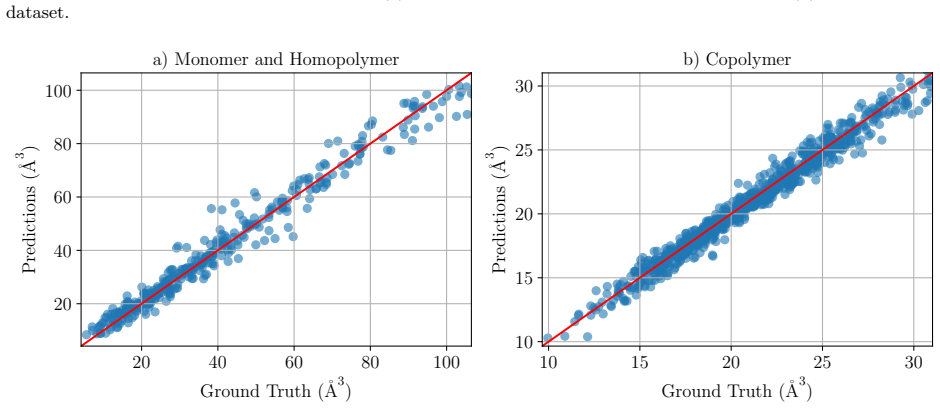
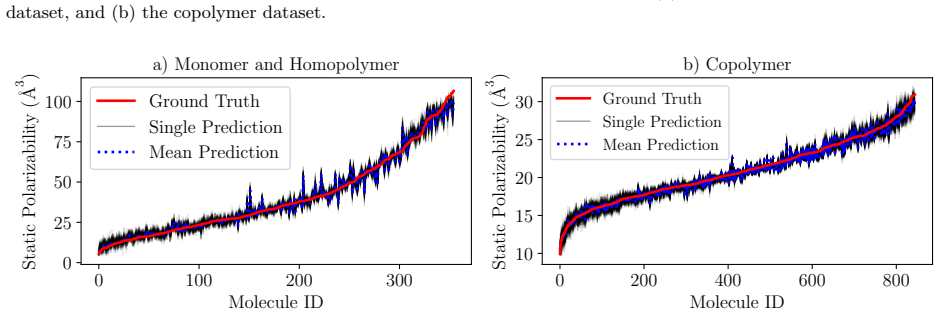
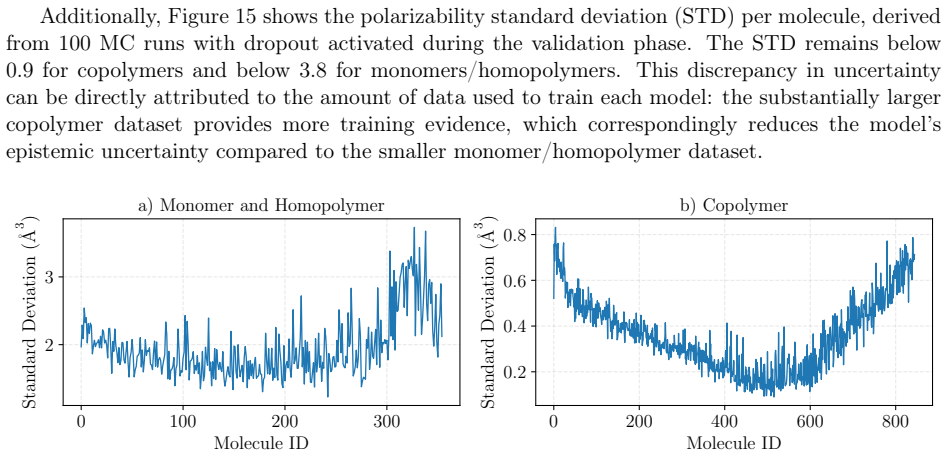
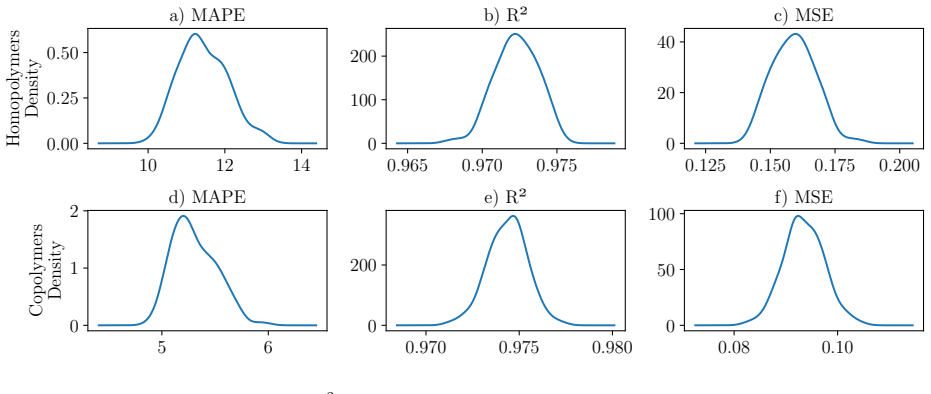
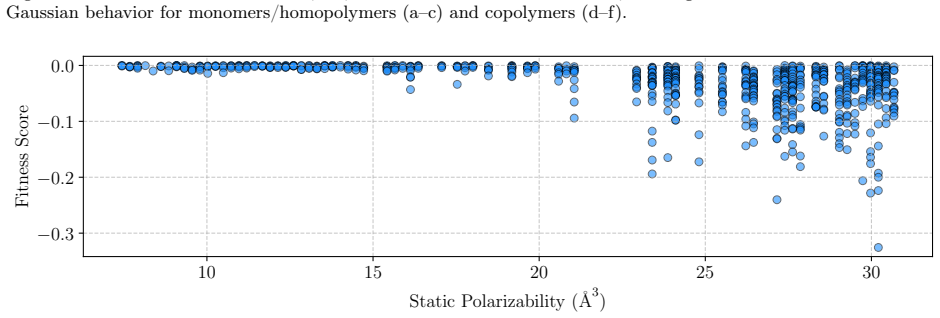
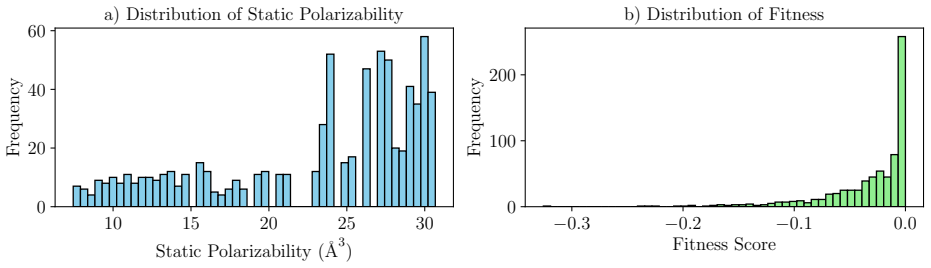
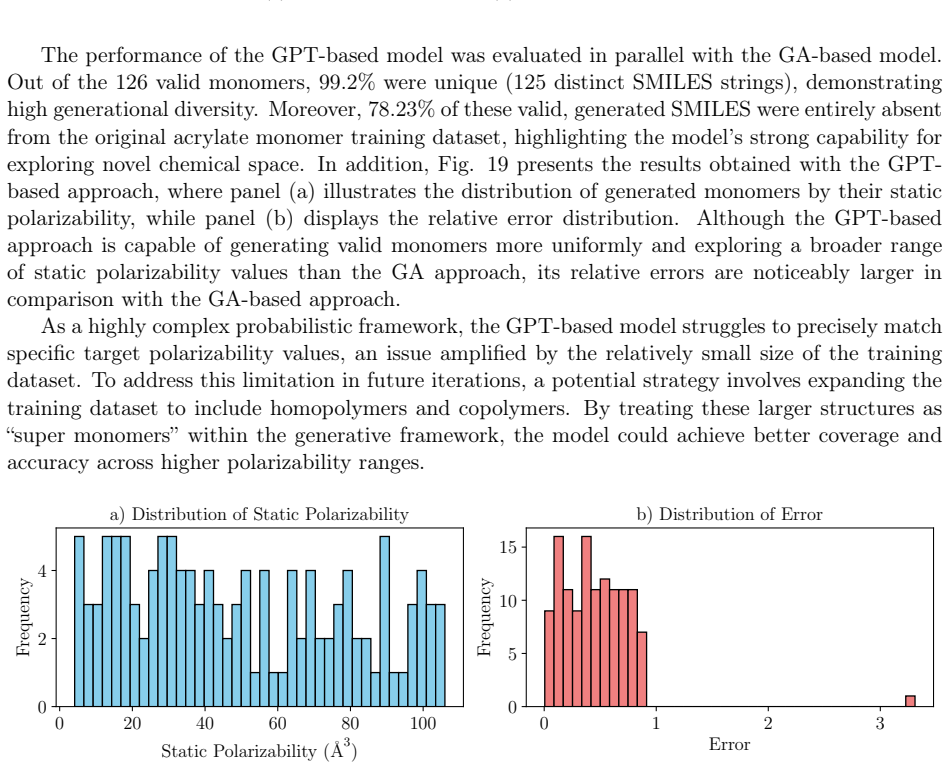
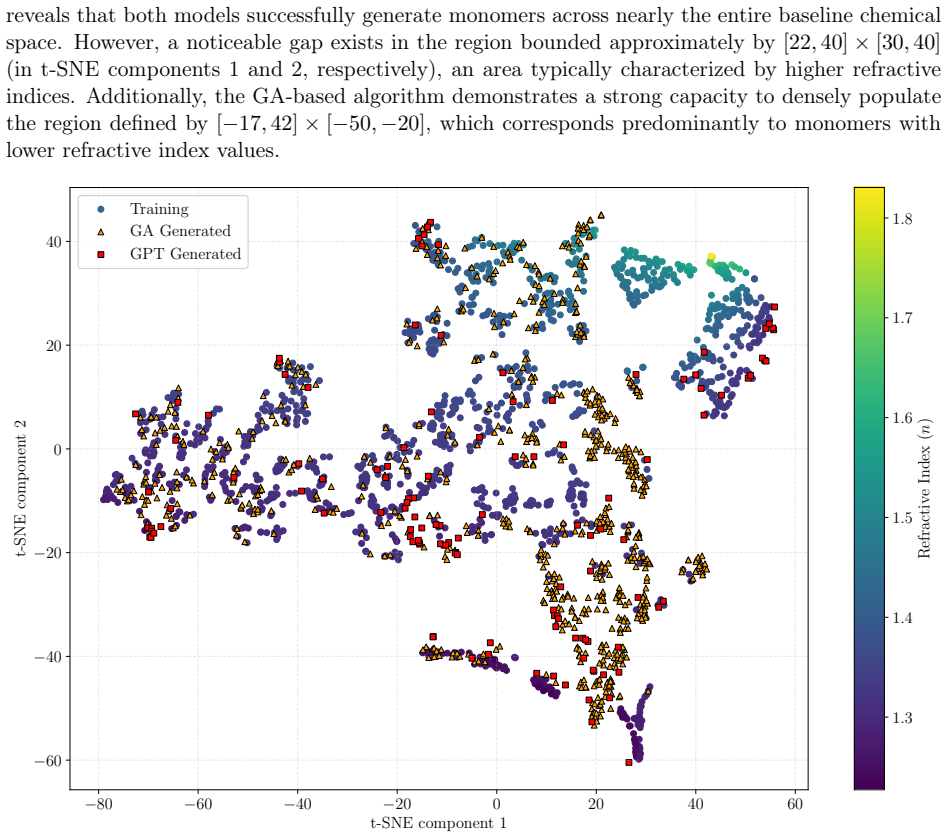
read the original abstract
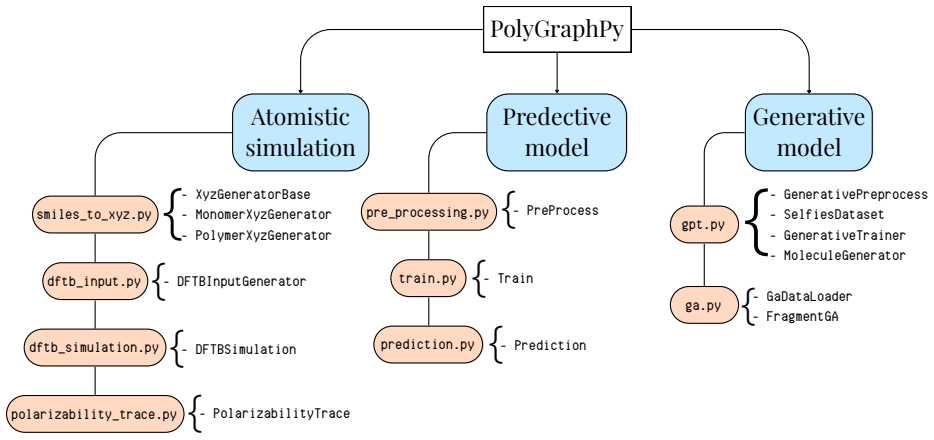
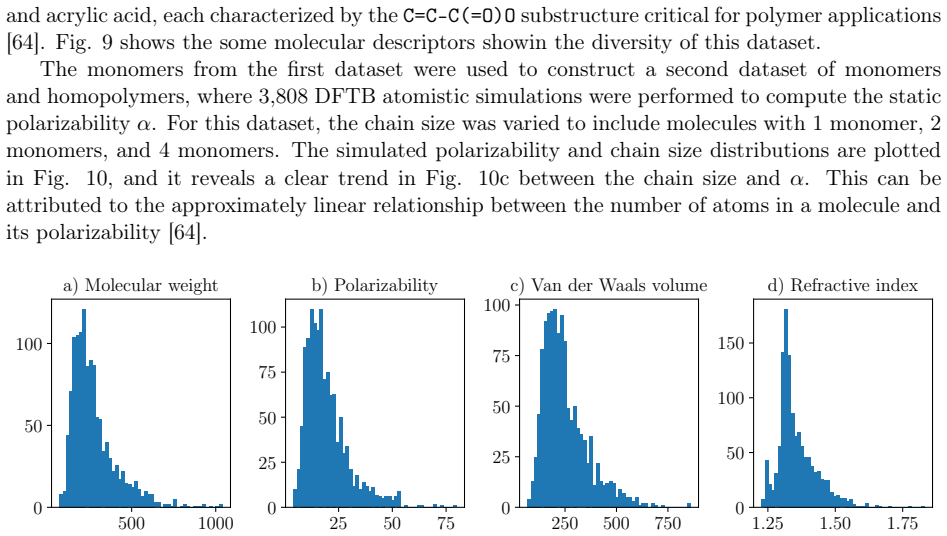
Polymers are indispensable materials with applications ranging from electronics to medicine owing to their versatility, which can be tailored by adjusting their chemical composition and architecture. The design space for these compounds is vast and governed by factors such as monomer classes, copolymer configurations (e.g., linear, branched, random, and alternating), chain size, stoichiometry, and material properties (e.g., density, refractive index, solubility, and Poisson's ratio). Exploring this space requires efficient computational methodologies for polymer science. To address this challenge, we introduce PolyGraphPy, an open-source Python framework that integrates atomistic simulations with machine learning for accurate property prediction and property-guided polymer design. The framework automates Density Functional Tight Binding calculations to efficiently construct structured datasets for monomers, homopolymers, and alternating copolymers. For property prediction, PolyGraphPy employs Bayesian Graph Neural Networks (GNNs) with stochastic graph representations to predict target properties, such as static polarizability, while providing robust uncertainty quantification. Furthermore, the platform incorporates two complementary generative models for the de novo design of targeted molecules: a SELFIES-based Generative Pretrained Transformer (GPT) and a Genetic Algorithm (GA) based on BRICS graph fragmentation. Demonstrated on a dataset of acrylates, PolyGraphPy provides a highly customizable end-to-end pipeline that reduces computational costs and accelerates data-driven polymer informatics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces PolyGraphPy, an open-source Python framework that automates DFTB calculations to build datasets for monomers, homopolymers, and copolymers; employs Bayesian GNNs with stochastic graph representations for property prediction (e.g., static polarizability) with uncertainty quantification; and integrates SELFIES-based GPT and BRICS-GA generative models for property-guided de novo polymer design. It claims this end-to-end pipeline, demonstrated on acrylates, reduces computational costs and accelerates polymer informatics.
Significance. A validated, integrated framework combining automated atomistic simulation, Bayesian GNN surrogates, and generative design could meaningfully lower barriers to polymer property prediction and inverse design. The open-source release and focus on customizable pipelines are positive features. However, the absence of any quantitative validation metrics means the significance cannot yet be assessed from the manuscript.
major comments (2)
- [Abstract / acrylates demonstration] Abstract and demonstration section: the central claim that Bayesian GNNs with stochastic graph representations deliver accurate predictions and robust UQ for properties such as static polarizability (required for property-guided design) is unsupported by any reported error metrics, calibration plots, comparison to DFTB/experiment, or ablation on the stochastic representation.
- [Abstract] Abstract: the assertion that the pipeline 'reduces computational costs and accelerates data-driven polymer informatics' lacks any supporting numbers (timings, dataset sizes, scaling comparisons, or baseline costs), which is load-bearing for the end-to-end utility claim.
minor comments (1)
- The manuscript should explicitly state code availability, installation instructions, and example notebooks to enable reproducibility of the claimed framework.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We agree that the current manuscript lacks the quantitative metrics needed to substantiate the central claims regarding prediction accuracy, uncertainty quantification, and computational efficiency. We will revise the manuscript to address these gaps directly. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [Abstract / acrylates demonstration] Abstract and demonstration section: the central claim that Bayesian GNNs with stochastic graph representations deliver accurate predictions and robust UQ for properties such as static polarizability (required for property-guided design) is unsupported by any reported error metrics, calibration plots, comparison to DFTB/experiment, or ablation on the stochastic representation.
Authors: We acknowledge that the submitted manuscript does not report error metrics (MAE, RMSE, R²), calibration plots, comparisons against DFTB or experimental values, or an ablation study isolating the stochastic graph representation. This is a genuine presentational gap that leaves the accuracy and UQ claims unsupported. In the revised manuscript we will add these elements to the demonstration section on acrylates, including tabulated performance numbers, reliability diagrams for the Bayesian predictions, and an explicit ablation comparing stochastic versus fixed graph inputs. revision: yes
-
Referee: [Abstract] Abstract: the assertion that the pipeline 'reduces computational costs and accelerates data-driven polymer informatics' lacks any supporting numbers (timings, dataset sizes, scaling comparisons, or baseline costs), which is load-bearing for the end-to-end utility claim.
Authors: We agree that the abstract claim is unsupported by any quantitative data in the current text. Although the framework description mentions automation of DFTB calculations, no timings, dataset cardinalities, or baseline comparisons appear. We will revise both the abstract and the methods/results sections to include concrete figures: number of monomers/homopolymers/copolymers generated, wall-clock times for the automated pipeline versus manual DFTB runs, and any scaling observations with system size. revision: yes
Circularity Check
No circularity: software framework paper with no derivation chain
full rationale
The manuscript describes an open-source Python framework (PolyGraphPy) that automates DFTB calculations, trains Bayesian GNNs on polymer graphs, and deploys generative models (SELFIES-GPT and BRICS-GA). No equations, uniqueness theorems, fitted-parameter predictions, or self-citation load-bearing arguments appear in the provided text. The central claims are engineering and demonstration statements about an end-to-end pipeline; they do not reduce to any input by construction. The paper is therefore self-contained as a tool description and receives the default non-circularity finding.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
S. Wu, H. Yamada, Y. Hayashi, M. Zamengo, R. Yoshida, Potentials and challenges of polymer informatics: exploiting machine learning for polymer design, 2020. URL:https://arxiv.org/ abs/2010.07683.arXiv:2010.07683
arXiv 2020
-
[2]
J. S. Peerless, N. J. B. Milliken, T. J. Oweida, M. D. Manning, Y. G. Yingling, Soft matter informatics: Current progress and challenges, Advanced Theory and Simulations 2 (2019) 1800129
2019
-
[3]
Weininger, Smiles, a chemical language and information system
D. Weininger, Smiles, a chemical language and information system. 1. introduction to method- ology and encoding rules, Journal of Chemical Information and Computer Sciences 28 (1988) 31–36
1988
-
[4]
T.-S. Lin, C. W. Coley, H. Mochigase, H. K. Beech, W. Wang, Z. Wang, E. Woods, S. L. Craig, J. A. Johnson, J. A. Kalow, K. F. Jensen, B. D. Olsen, Bigsmiles: A structurally-based line notation for describing macromolecules, ACS Central Science 5 (2019) 1523–1531. PMID: 31572779
2019
-
[5]
S. R. Heller, A. McNaught, I. Pletnev, S. Stein, D. Tchekhovskoi, Inchi, the iupac international chemical identifier, Journal of Cheminformatics 7 (2015) 23
2015
-
[6]
M. Guo, W. Shou, L. Makatura, T. Erps, M. Foshey, W. Matusik, Polygrammar: Grammar for digital polymer representation and generation, Advanced Science 9 (2022) 2101864
2022
-
[7]
Krenn, F
M. Krenn, F. Häse, A. Nigam, P. Friederich, A. Aspuru-Guzik, Self-referencing embedded strings (selfies): A 100% robust molecular string representation, Machine Learning: Science and Technology 1 (2020) 045024
2020
-
[8]
Aldeghi, C
M. Aldeghi, C. W. Coley, A graph representation of molecular ensembles for polymer property prediction, Chemical Science 13 (2022) 10486–10498
2022
-
[9]
C. Kim, A. Chandrasekaran, T. D. Huan, D. Das, R. Ramprasad, Polymer genome: A data- powered polymer informatics platform for property predictions, The Journal of Physical Chem- istry C 122 (2018) 17575–17585
2018
-
[10]
Doan Tran, C
H. Doan Tran, C. Kim, L. Chen, A. Chandrasekaran, R. Batra, S. Venkatram, D. Kamal, J. P. Lightstone, R. Gurnani, P. Shetty, M. Ramprasad, J. Laws, M. Shelton, R. Ramprasad, Machine-learning predictions of polymer properties with polymer genome, Journal of Applied Physics 128 (2020) 171104
2020
-
[11]
K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen, R. Barzilay, Analyzing learned molecular representations for property prediction, Journal of Chemical Information and Modeling 59 (2019) 3370–3388. PMID: 31361484
2019
-
[12]
E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green, C. J. McGill, Chemprop: A machine learning package for chemical property prediction, Journal of Chemical Information and Modeling 64 (2024) 9–17. PMID: 38147829
2024
-
[13]
Ignacz, M
G. Ignacz, M. I. Baig, K. Gopalsamy, A. Villa, S. Nunes, B. Ghanem, T. Shastry, S. K. Kumar, G.Szekely, Adata-drivenapproachtointerfacialpolymerizationexploitingmachinelearningfor predicting thin-film composite membrane formation, Materials Horizons 12 (2025) 9009–9025. 23
2025
-
[14]
S. Sun, F. Tian, C. Zhao, M. Xie, W. Li, W. Yu, K. Cui, L. Li, Directed message passing neural networks enhanced graph convolutional learning for accurate polymer density prediction, The Journal of Chemical Physics 163 (2025)
2025
-
[15]
Correia, J
J. Correia, J. Capela, M. Rocha, Deepmol: an automated machine and deep learning framework for computational chemistry, Journal of Cheminformatics 16 (2024) 136
2024
-
[16]
Bicerano, D
J. Bicerano, D. Rigby, C. Freeman, B. LeBlanc, J. Aubry, Polymer expert – a software tool for de novo polymer design, Computational Materials Science 235 (2024) 112810
2024
-
[17]
Nanjo, Arifin, H
S. Nanjo, Arifin, H. Maeda, Y. Hayashi, K. Hatakeyama-Sato, R. Himeno, T. Hayakawa, R. Yoshida, Spacier: On-demand polymer design with fully automated all-atom classical molecular dynamics integrated into machine learning pipelines, npj Computational Materi- als 11 (2025) 16
2025
-
[18]
I. Priyadarsini, S. Takeda, L. Hamada, E. V. Brazil, E. Soares, H. Shinohara, Self-bart: A transformer-based molecular representation model using selfies, 2024. URL:https://arxiv. org/abs/2410.12348.arXiv:2410.12348
arXiv 2024
-
[19]
M. Lewis, Y. Liu, N. Goyal, M. Ghazvininejad, A. Mohamed, O. Levy, V. Stoyanov, L. Zettlemoyer, Bart: Denoising sequence-to-sequence pre-training for natural language gen- eration, translation, and comprehension, 2019. URL:https://arxiv.org/abs/1910.13461. arXiv:1910.13461
Pith/arXiv arXiv 2019
-
[20]
H. Kim, M. Kim, S. Choi, J. Park, Genetic-guided gflownets for sample efficient molecular optimization, 2024. URL:https://arxiv.org/abs/2402.05961.arXiv:2402.05961
arXiv 2024
-
[21]
Bongini, M
P. Bongini, M. Bianchini, F. Scarselli, Molecular generative graph neural networks for drug discovery, Neurocomputing 450 (2021) 242–252
2021
-
[22]
Elstner, Scc-dftb: What is the proper degree of self-consistency?, The Journal of Physical Chemistry A 111 (2007) 5614–5621
M. Elstner, Scc-dftb: What is the proper degree of self-consistency?, The Journal of Physical Chemistry A 111 (2007) 5614–5621. PMID: 17564420
2007
-
[23]
Hourahine, B
B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. P...
2020
-
[24]
Radford, J
A. Radford, J. Wu, R. Child, D. Luan, D. Amodei, I. Sutskever, Language models are unsu- pervised multitask learners, OpenAI (2019). Accessed: 2024-11-15
2019
-
[25]
J. H. Holland, Genetic algorithms, Scientific American 267 (1992) 66–73
1992
-
[26]
Katoch, S
S. Katoch, S. S. Chauhan, V. Kumar, A review on genetic algorithm: past, present, and future, Multimedia Tools and Applications 80 (2021) 8091–8126
2021
-
[27]
Scarselli, M
F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner, G. Monfardini, The graph neural network model, IEEE Transactions on Neural Networks 20 (2009) 61–80. 24
2009
-
[28]
T. N. Kipf, M. Welling, Variational graph auto-encoders, 2016. URL:https://arxiv.org/ abs/1611.07308.arXiv:1611.07308
Pith/arXiv arXiv 2016
-
[29]
H. Gao, S. Ji, Graph u-nets, 2019. URL:https://arxiv.org/abs/1905.05178. arXiv:1905.05178
Pith/arXiv arXiv 2019
-
[30]
Degen, C
J. Degen, C. Wegscheid-Gerlach, A. Zaliani, M. Rarey, On the art of compiling and using ’drug-like’ chemical fragment spaces, ChemMedChem 3 (2008) 1503–1507
2008
-
[31]
A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Köpf, E. Yang, Z. DeVito, M. Raison, A. Te- jani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai, S. Chintala, Pytorch: An imperative style, high-performance deep learning library, 2019. URL:https://arxiv.org/abs/1912.01703. arXiv:...
Pith/arXiv arXiv 2019
-
[32]
M. Fey, J. E. Lenssen, Fast graph representation learning with pytorch geometric, 2019. URL: https://arxiv.org/abs/1903.02428.arXiv:1903.02428
Pith/arXiv arXiv 2019
-
[33]
Accessed: 2025- 07-12
DFTB+ Development Team, DFTB+: A software package for efficient approximate density functional theory based atomistic simulations,https://dftbplus.org, 2025. Accessed: 2025- 07-12
2025
-
[34]
Hourahine, M
B. Hourahine, M. Berdakin, J. A. Bich, F. P. Bonafé, C. Camacho, Q. Cui, M. Y. Deshaye, G. Díaz Mirón, S. Ehlert, M. Elstner, T. Frauenheim, N. Goldman, R. A. González León, T. van der Heide, S. Irle, T. Kowalczyk, T. Kubař, I. S. Lee, C. R. Lien-Medrano, A. Maryewski, T. Melson, S. K. Min, T. Niehaus, A. M. N. Niklasson, A. Pecchia, K. Reuter, C. G. Sánc...
2025
-
[35]
Hohenberg, W
P. Hohenberg, W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136 (1964) B864–B871
1964
-
[36]
W. Kohn, L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev. 140 (1965) A1133–A1138
1965
-
[37]
S. U. Patil, M. S. Radue, W. A. Pisani, P. Deshpande, H. Xu, H. Al Mahmud, T. Dumitrică, G. M. Odegard, Interfacial characteristics between flattened cnt stacks and polyimides: A molecular dynamics study, Computational Materials Science 185 (2020) 109970
2020
-
[38]
Elstner, T
M. Elstner, T. Frauenheim, E. Kaxiras, G. Seifert, S. Suhai, A self-consistent charge density- functional based tight-binding scheme for large biomolecules, physica status solidi (b) 217 (2000) 357–376
2000
-
[39]
M. Gaus, X. Lu, M. Elstner, Q. Cui, Parameterization of dftb3/3ob for sulfur and phosphorus for chemical and biological applications, Journal of Chemical Theory and Computation 10 (2014) 1518–1537. PMID: 24803865
2014
-
[40]
J. C. Slater, G. F. Koster, Simplified lcao method for the periodic potential problem, Physical Review 94 (1954) 1498–1524
1954
-
[41]
Turcani, E
L. Turcani, E. Berardo, K. E. Jelfs, stk: A python toolkit for supramolecular assembly, Journal of Computational Chemistry 39 (2018) 1456–1465. 25
2018
-
[42]
M. B. Oviedo, C. F. A. Negre, C. G. Sánchez, Dynamical simulation of the optical response of photosynthetic pigments, Phys. Chem. Chem. Phys. 12 (2010) 6706–6711
2010
-
[43]
Kearnes, K
S. Kearnes, K. McCloskey, M. Berndl, V. Pande, P. Riley, Molecular graph convolutions: Moving beyond fingerprints, Journal of Computer-Aided Molecular Design 30 (2016) 595–608
2016
-
[44]
J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackermann, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay, J. J. Collins, A deep learning approach to antibiotic discovery, Cell 180 (2020) 688–702.e13
2020
-
[45]
Jiang, Z
D. Jiang, Z. Wu, C.-Y. Hsieh, G. Chen, B. Liao, Z. Wang, C. Shen, D. Cao, J. Wu, T. Hou, Could graph neural networks learn better molecular representation for drug discovery? a com- parison study of descriptor-based and graph-based models, Journal of Cheminformatics 13 (2021) 12
2021
-
[46]
M. Zeng, J. N. Kumar, Z. Zeng, R. Savitha, V. R. Chandrasekhar, K. Hippalgaonkar, Graph convolutional neural networks for polymers property prediction, 2018. URL:https://arxiv. org/abs/1811.06231.arXiv:1811.06231
Pith/arXiv arXiv 2018
-
[47]
Gurnani, C
R. Gurnani, C. Kuenneth, A. Toland, R. Ramprasad, Polymer informatics at scale with mul- titask graph neural networks, Chemistry of Materials 35 (2023) 1560–1567
2023
-
[48]
F. Wang, W. Guo, M. Cheng, S. Yuan, H. Xu, Z. Gao, Mmpolymer: A multimodal multitask pretraining framework for polymer property prediction, 2024. URL:https://arxiv.org/abs/ 2406.04727.arXiv:2406.04727
arXiv 2024
-
[49]
T. N. Kipf, M. Welling, Semi-supervised classification with graph convolutional networks, 2017. URL:https://arxiv.org/abs/1609.02907.arXiv:1609.02907
Pith/arXiv arXiv 2017
-
[50]
Wasserman, Bayesian model selection and model averaging, Journal of Mathematical Psychology 44 (2000) 92–107
L. Wasserman, Bayesian model selection and model averaging, Journal of Mathematical Psychology 44 (2000) 92–107
2000
-
[51]
D. M. Blei, A. Kucukelbir, J. D. McAuliffe, Variational inference: A review for statisticians, Journal of the American Statistical Association 112 (2017) 859–877
2017
-
[52]
Y. Gal, Z. Ghahramani, Bayesian convolutional neural networks with bernoulli approximate variational inference, 2015. URL:https://arxiv.org/abs/1506.02158. doi:10.48550/ARXIV. 1506.02158
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv 2015
-
[53]
L. V. Jospin, H. Laga, F. Boussaid, W. Buntine, M. Bennamoun, Hands-on bayesian neural networks—a tutorial for deep learning users, IEEE Computational Intelligence Magazine 17 (2022) 29–48
2022
-
[54]
O. Ronneberger, P. Fischer, T. Brox, U-net: Convolutional networks for biomedical image segmentation, 2015. URL:https://arxiv.org/abs/1505.04597.arXiv:1505.04597
Pith/arXiv arXiv 2015
-
[55]
A.Vaswani, N.Shazeer, N.Parmar, J.Uszoreit, L.Jones, A.N.Gomez, L.Kaiser, I.Polosukhin, Attention is all you need, Advances in Neural Information Processing Systems 30 (2017) 5998– 6008
2017
-
[56]
Bilodeau, W
C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay, K. F. Jensen, Generative models for molecular discovery: Recent advances and challenges, Wiley Interdisciplinary Reviews: Computational Molecular Science 12 (2022) e1608. 26
2022
-
[57]
N. C. Frey, V. Gadepally, S. Samsi, A. Speth, B. Subramanian, Scalable generative models for molecular design, Journal of Chemical Information and Modeling 63 (2023) 1905–1915
2023
-
[58]
Nigam, R
A. Nigam, R. Pollice, M. F. D. Hurley, R. J. Hickman, M. Aldeghi, N. Yoshikawa, S. Chithrananda, A. Aspuru-Guzik, Artificial intelligence in chemistry: Current trends and future directions, Journal of Chemical Information and Modeling 60 (2020) 6025–6041
2020
-
[59]
J. H. Jensen, A graph-based genetic algorithm and generative model/monte carlo tree search for the exploration of chemical space, Chemical Science 10 (2019) 3567–3572
2019
-
[60]
Available athttps://huggingface.co/openai-community/gpt2
Hugging Face, Gpt-2: Language models are unsupervised multitask learners, Hugging Face Model Hub, 2019. Available athttps://huggingface.co/openai-community/gpt2
2019
-
[61]
I. Loshchilov, F. Hutter, Decoupled weight decay regularization, 2019. URL:https://arxiv. org/abs/1711.05101.arXiv:1711.05101
Pith/arXiv arXiv 2019
-
[62]
S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang, E. E. Bolton, Pubchem in 2021: New data content and improved web interfaces, Nucleic Acids Research 49 (2021) D1388–D1395
2021
-
[63]
nlm.nih.gov/, 2025
National Center for Biotechnology Information (NCBI), Pubchem,https://pubchem.ncbi. nlm.nih.gov/, 2025. Accessed: [Current Date]
2025
-
[64]
R. J. Young, P. A. Lovell, Introduction to polymers, Chapman and Hall (1999)
1999
-
[65]
Y. Gal, Z. Ghahramani, Dropout as a bayesian approximation: Representing model uncertainty in deep learning, 2016. URL:https://arxiv.org/abs/1506.02142.arXiv:1506.02142
Pith/arXiv arXiv 2016
-
[66]
Y. Gal, Z. Ghahramani, Dropout as a bayesian approximation: Appendix, 2016. URL:https: //arxiv.org/abs/1506.02157.arXiv:1506.02157
Pith/arXiv arXiv 2016
-
[67]
van der Maaten, G
L. van der Maaten, G. Hinton, Visualizing data using t-sne, Journal of Machine Learning Research 9 (2008) 2579–2605. 27
2008
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.