QmDFT for Polycyclic Aromatics: Balancing Embedding Ground-State Fidelity and Experimental Gap Estimation
Pith reviewed 2026-06-30 07:41 UTC · model grok-4.3
The pith
An adaptive damping and DIIS protocol stabilizes QmDFT embedding, enabling hybrid functionals to estimate experimental E0-0 gaps in PAHs from ground-state calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
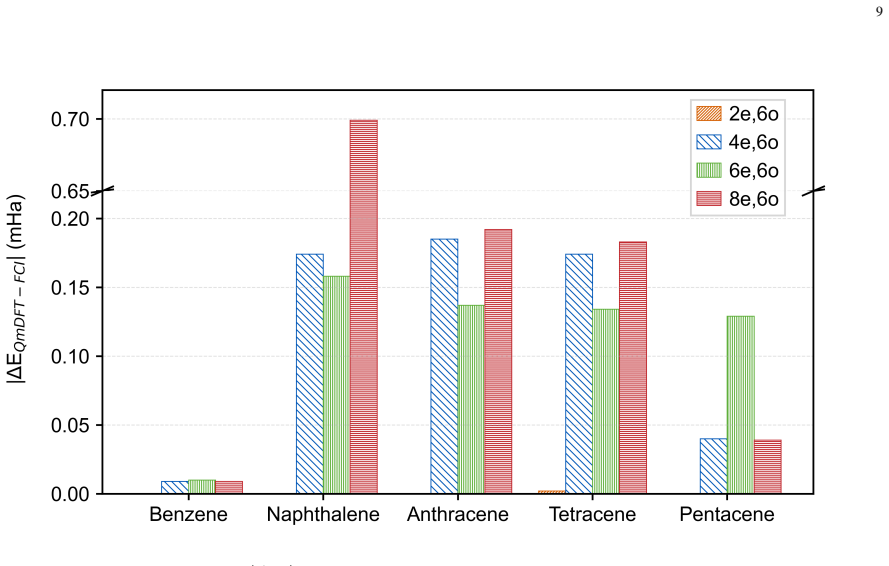
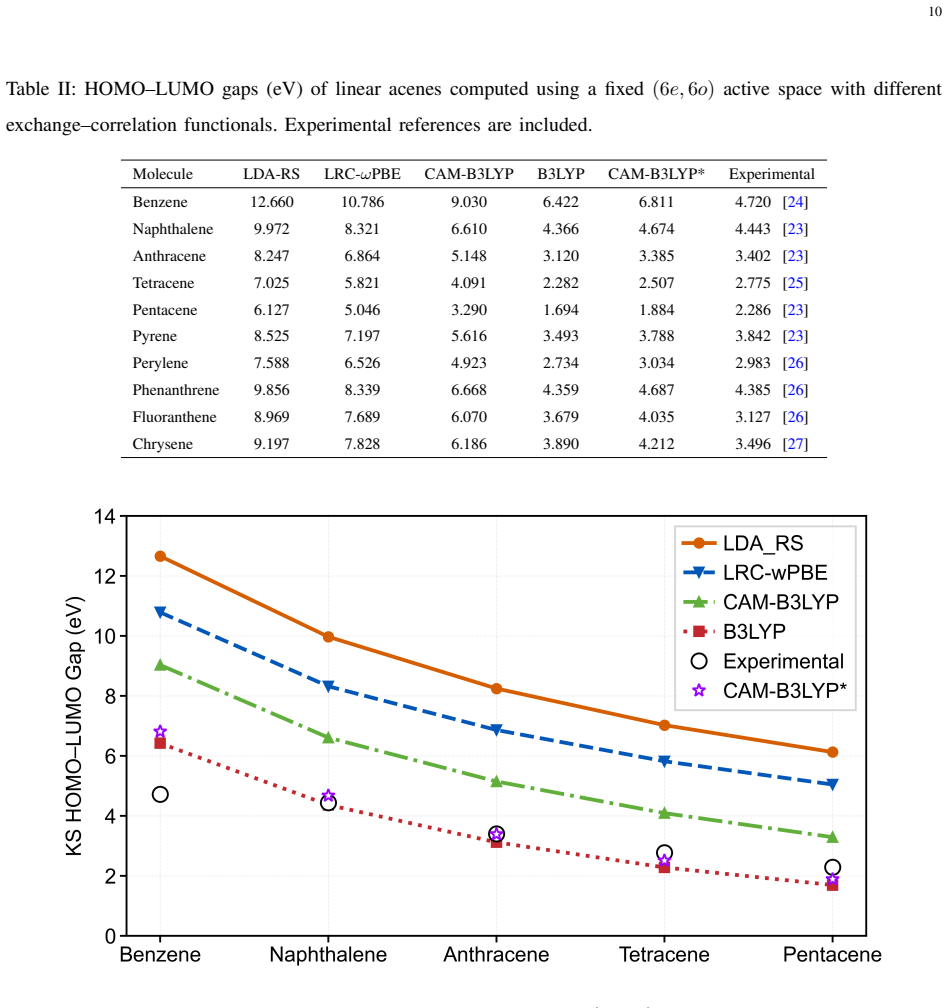
The adaptive damping and direct inversion in the iterative subspace accelerated protocol stabilizes the QmDFT embedding procedure, enabling robust integration of hybrid functionals such as B3LYP and CAM-B3LYP. On the benchmark set of ten PAHs, LDA yields near-quantitative agreement with FCI reference energies and thermochemical isomerization data, while B3LYP delivers significantly improved agreement with experimental E0-0 transition values. The resulting mapping from ground-state properties to E0-0 values bypasses explicit excited-state calculations and reduces computational overhead.
What carries the argument
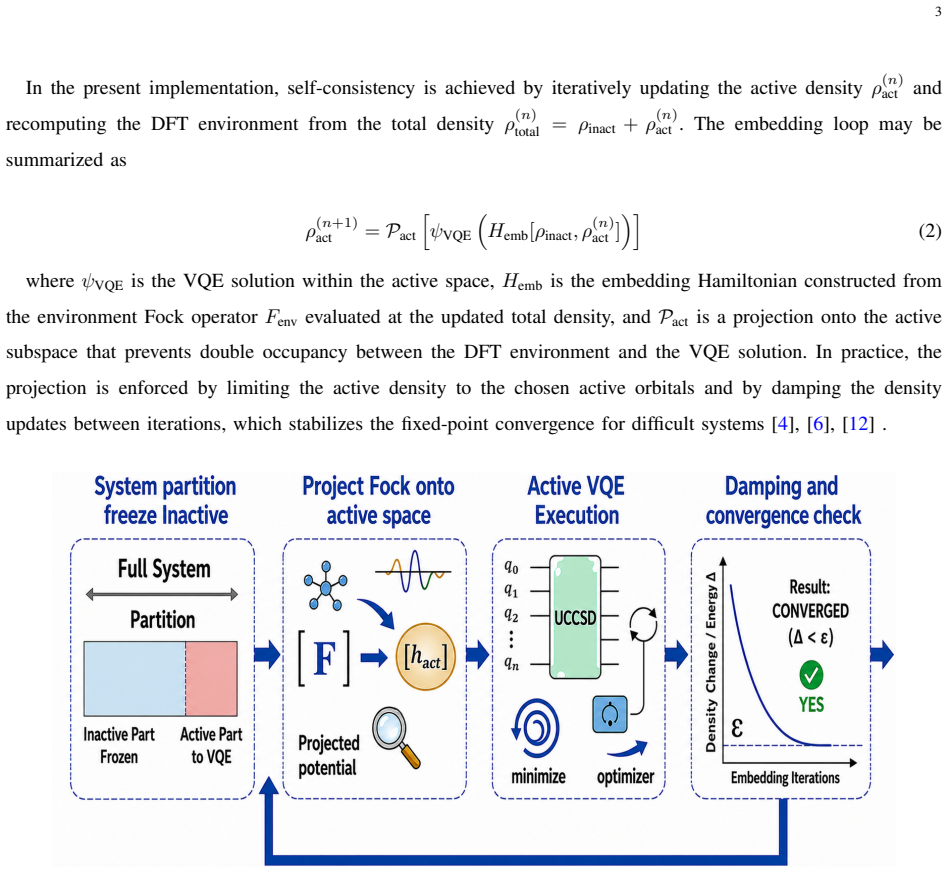
Adaptive damping and DIIS accelerated protocol that stabilizes the embedding cycle in QmDFT, allowing hybrid functionals to converge reliably.
If this is right
- B3LYP in the stabilized embedding yields significantly improved agreement with experimental E0-0 transition values.
- CAM-B3LYP provides balanced overall performance among the hybrid functionals screened.
- LDA approaches achieve near-quantitative agreement with FCI reference energies and thermochemical isomerization benchmarks.
- Ground-state results map directly to E0-0 values, bypassing explicit excited-state calculations.
- The framework supplies guidance for functional selection in quantum embedding studies of PAHs and related pi-conjugated materials.
Where Pith is reading between the lines
- The stabilization method could extend to larger pi-conjugated systems where convergence instabilities are the main barrier.
- The ground-state to E0-0 mapping may simplify screening workflows for other low-dimensional conjugated materials.
- Similar damping and DIIS protocols could address functional instability in alternative embedding schemes.
- Trends observed here suggest testing B3LYP first for gap estimation tasks on untested molecular sizes or topologies.
Load-bearing premise
The functional-dependent performance seen on the ten selected linear and fused PAH molecules will hold for larger or differently structured pi-conjugated systems.
What would settle it
A high-level calculation or measured E0-0 value for a PAH outside the ten-molecule benchmark set that deviates substantially from the stabilized B3LYP QmDFT prediction.
Figures
read the original abstract
Quantum Embedding density functional theory (QmDFT) embedding offers a highly scalable approach to improve treatment for large highly correlated pi conjugated systems. However, estimating advanced electronic structure properties in polycyclic aromatic hydrocarbons (PAHs) needs advanced exchange correlation functionals that frequently trigger convergence instabilities during the embedding cycle. In this work, we introduce an adaptive damping and direct inversion in the iterative subspace (DIIS) accelerated protocol that stabilizes the embedding procedure, enabling robust integration of hybrid functionals like B3LYP and CAM B3LYP. Using 10 selected PAHs (linear and fused) molecules as a benchmark. We demonstrate a clear functional-dependent ground-state energetics and frontier-orbital gap estimation. While LDA based approaches yield near-quantitative agreement with FCI in DFT reference energies and is further supported by thermochemical isomerization benchmarks, while B3LYP provide significantly improved agreement with experimental E0-0 transition values. This mapping allows us to bypass explicit excited-state calculations for E0 0 values, thereby significantly reducing computational overhead. Among the hybrid functionals screened, CAM B3LYP offers a balanced overall performance. Our results establish a stable QmDFT framework and provide useful guidance for functional selection for quantum embedding studies of PAHs and related low-dimensional pi-conjugated materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces an adaptive damping and DIIS-accelerated protocol to stabilize QmDFT embedding calculations when incorporating hybrid functionals for polycyclic aromatic hydrocarbons. On a benchmark set of 10 linear and fused PAH molecules, LDA embedding is reported to achieve near-quantitative agreement with FCI reference energies and thermochemical isomerization benchmarks, while B3LYP embedding yields improved numerical agreement with experimental E0-0 transition energies via ground-state frontier orbital gaps, thereby permitting bypass of explicit excited-state calculations. CAM-B3LYP is identified as providing balanced overall performance.
Significance. If the reported functional-dependent correlations and stabilization protocol prove robust beyond the current benchmark, the approach could supply a scalable route to ground-state-based gap estimates for larger pi-conjugated systems, reducing reliance on excited-state methods while supplying practical functional-selection guidance for embedding studies of PAHs and related materials.
major comments (3)
- [Abstract] Abstract: The headline claim that B3LYP (or CAM-B3LYP) ground-state frontier gaps inside the stabilized embedding quantitatively track experimental E0-0 transitions rests solely on an observed numerical correlation for exactly ten linear/fused PAHs; no physical derivation, error-cancellation argument, or validation on additional systems is supplied, rendering the bypass of excited-state calculations an empirical finding whose generality is untested.
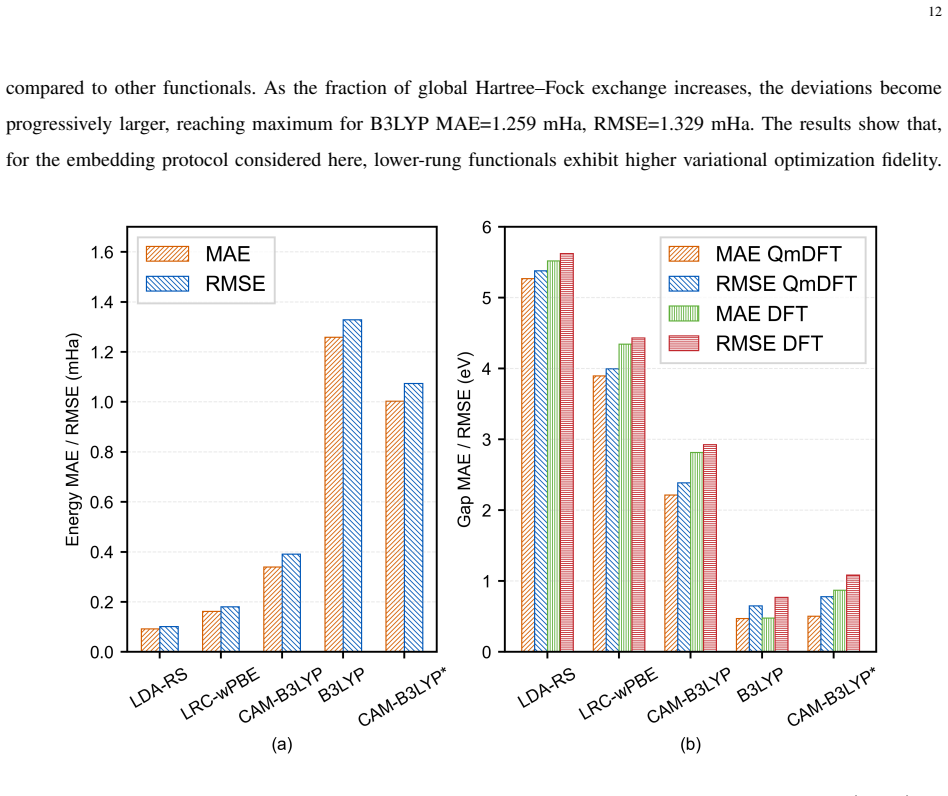
- [Benchmark section] Benchmark section: No error bars, convergence statistics, or explicit criteria for selecting the ten molecules are reported, and the functional-dependent performance trends (LDA vs. B3LYP) are presented without statistical quantification of the correlation strength, which is load-bearing for the central mapping claim.
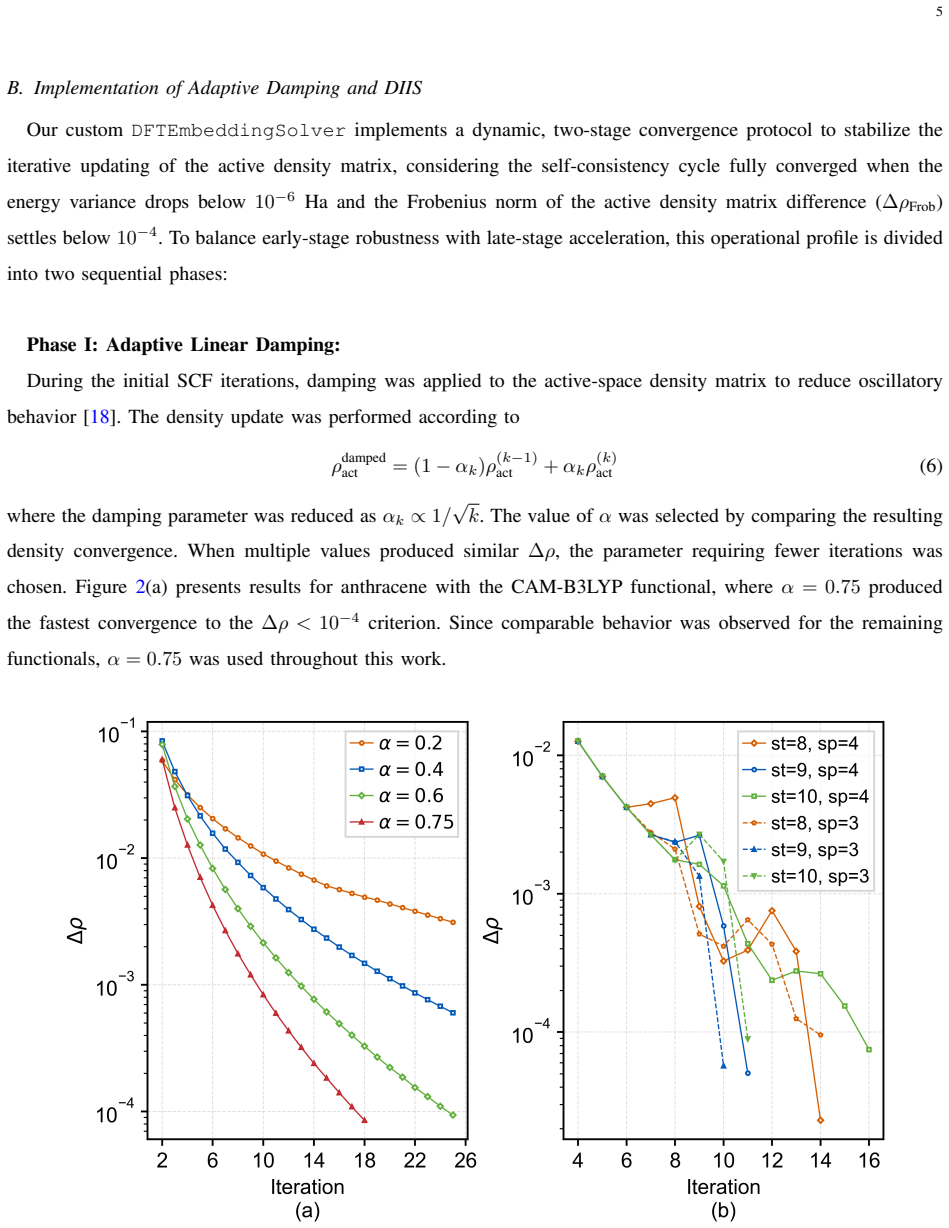
- [Methods] Methods / protocol description: The adaptive damping parameters are free parameters whose values enable convergence for hybrids, yet no sensitivity study or explicit demonstration of how the damping/DIIS combination removes instabilities for B3LYP/CAM-B3LYP is provided, leaving the robustness of the stabilization protocol incompletely documented.
minor comments (2)
- [Abstract] Abstract: Inconsistent notation ('CAM B3LYP', 'E0 0', 'E0-0 values') and awkward sentence structure in the LDA/B3LYP comparison clause require correction for clarity.
- Throughout: Several instances of missing hyphenation in compound terms and minor grammatical issues in the results narrative should be addressed in copy-editing.
Simulated Author's Rebuttal
We thank the referee for the insightful comments on our manuscript. We address each major comment below and indicate the revisions we will make to improve clarity and documentation.
read point-by-point responses
-
Referee: [Abstract] The headline claim that B3LYP (or CAM-B3LYP) ground-state frontier gaps inside the stabilized embedding quantitatively track experimental E0-0 transitions rests solely on an observed numerical correlation for exactly ten linear/fused PAHs; no physical derivation, error-cancellation argument, or validation on additional systems is supplied, rendering the bypass of excited-state calculations an empirical finding whose generality is untested.
Authors: The manuscript presents the agreement as a numerical correlation observed for the benchmark set, without claiming a fundamental physical derivation. This empirical finding is useful for the systems studied and provides practical guidance for functional choice. We will revise the abstract to more clearly indicate that the bypass is based on the observed correlation for these 10 PAHs and note the empirical nature, suggesting further testing for generality. revision: partial
-
Referee: [Benchmark section] No error bars, convergence statistics, or explicit criteria for selecting the ten molecules are reported, and the functional-dependent performance trends (LDA vs. B3LYP) are presented without statistical quantification of the correlation strength, which is load-bearing for the central mapping claim.
Authors: We will include explicit selection criteria for the 10 PAHs (representative linear and fused structures with available reference data). Additionally, we will report mean absolute errors, maximum deviations, and correlation coefficients (e.g., R-squared values) to quantify the trends between functionals and references. Error bars will be added where multiple calculations allow, though the calculations are deterministic. revision: yes
-
Referee: [Methods] The adaptive damping parameters are free parameters whose values enable convergence for hybrids, yet no sensitivity study or explicit demonstration of how the damping/DIIS combination removes instabilities for B3LYP/CAM-B3LYP is provided, leaving the robustness of the stabilization protocol incompletely documented.
Authors: The protocol description includes the adaptive damping and DIIS to achieve convergence where standard SCF fails for hybrids. To better document robustness, we will add a supplementary note or figure showing convergence behavior with and without the damping/DIIS for a representative B3LYP case on one PAH, illustrating the stabilization effect. revision: partial
Circularity Check
No significant circularity; results rest on external benchmarks
full rationale
The paper reports numerical outcomes from QmDFT embedding on a fixed set of 10 PAHs, with LDA energies compared to external FCI references and B3LYP gaps compared to experimental E0-0 values. The adaptive damping/DIIS protocol is introduced as a stabilization technique, but no equation or claim reduces a target quantity to a parameter fitted from that same quantity. No self-citations are invoked as load-bearing uniqueness theorems, and the functional-dependent agreement is presented as an empirical observation rather than a constructed identity. The derivation chain is therefore self-contained against independent data.
Axiom & Free-Parameter Ledger
free parameters (1)
- adaptive damping parameters
axioms (1)
- domain assumption Standard Kohn-Sham DFT with chosen exchange-correlation functionals yields meaningful ground-state energies and orbital gaps for the embedded system.
Reference graph
Works this paper leans on
-
[1]
The radical character of the acenes: A density matrix renormalization group study,
J. Hachmann, J. J. Dorando, M. Avil ´es, and G. K.-L. Chan, “The radical character of the acenes: A density matrix renormalization group study,”The Journal of Chemical Physics, vol. 127, no. 13, p. 134309, 10 2007. [Online]. Available: https://doi.org/10.1063/1.2768362
-
[2]
Excitation gaps of finite-sized systems from optimally tuned range-separated hybrid functionals,
L. Kronik, T. Stein, S. Refaely-Abramson, and R. Baer, “Excitation gaps of finite-sized systems from optimally tuned range-separated hybrid functionals,”Journal of Chemical Theory and Computation, vol. 8, no. 5, pp. 1515–1531, 2012, pMID: 26593646. [Online]. Available: https://doi.org/10.1021/ct2009363
-
[3]
Range separation and local hybridization in density functional theory,
T. M. Henderson, B. G. Janesko, and G. E. Scuseria, “Range separation and local hybridization in density functional theory,”The Journal of Physical Chemistry A, vol. 112, no. 49, pp. 12 530–12 542, 2008, pMID: 19006280. [Online]. Available: https://doi.org/10.1021/jp806573k
-
[4]
A simple, exact density-functional-theory embedding scheme,
F. R. Manby, M. Stella, J. D. Goodpaster, and T. F. Miller, III, “A simple, exact density-functional-theory embedding scheme,”J. Chem. Theory Comput., vol. 8, pp. 2564–2568, Aug 2012. [Online]. Available: https://doi.org/10.1021/ct300544e
-
[5]
Density matrix embedding: A strong-coupling quantum embedding theory,
G. Knizia and G. K.-L. Chan, “Density matrix embedding: A strong-coupling quantum embedding theory,”Journal of Chemical Theory and Computation, vol. 9, no. 3, p. 1428–1432, Feb. 2013. [Online]. Available: http://dx.doi.org/10.1021/ct301044e
-
[6]
Quantum embedding method for the simulation of strongly correlated systems on quantum computers,
M. Rossmannek, F. Pavo ˇsevi´c, A. Rubio, and I. Tavernelli, “Quantum embedding method for the simulation of strongly correlated systems on quantum computers,”The Journal of Physical Chemistry Letters, vol. 14, no. 14, p. 3491–3497, Apr. 2023. [Online]. Available: http://dx.doi.org/10.1021/acs.jpclett.3c00330
-
[7]
Scalable quantum-classical dft embedding for nisq molecular simulation,
N. Manglani, S. K. Maity, R. Thapa, and S. Wandhekar, “Scalable quantum-classical dft embedding for nisq molecular simulation,” 2026. [Online]. Available: https://arxiv.org/abs/2602.01994
-
[8]
Physical Review Letters , volume =
J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approximation made simple,”Phys. Rev. Lett., vol. 77, pp. 3865–3868, Oct 1996. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevLett.77.3865
-
[9]
Density-functional exchange-energy approximation with correct asymptotic behavior,
A. D. Becke, “Density-functional exchange-energy approximation with correct asymptotic behavior,”Phys. Rev. A, vol. 38, pp. 3098–3100, Sep 1988. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevA.38.3098
-
[10]
Density-functional thermochemistry. iii. the role of exact exchange,
A. D. Becke, “Density-functional thermochemistry. iii. the role of exact exchange,”The Journal of Chemical Physics, vol. 98, no. 7, pp. 5648–5652, 04 1993. [Online]. Available: https://doi.org/10.1063/1.464913
-
[11]
A new hybrid exchange–correlation functional using the coulomb-attenuating method (cam-b3lyp),
T. Yanai, D. P. Tew, and N. C. Handy, “A new hybrid exchange–correlation functional using the coulomb-attenuating method (cam-b3lyp),”Chemical Physics Letters, vol. 393, no. 1, pp. 51–57, 2004. [Online]. Available: https://www.sciencedirect.com/science/ article/pii/S0009261404008620
2004
-
[12]
M. Rossmannek, P. K. Barkoutsos, P. J. Ollitrault, and I. Tavernelli, “Quantum hf/dft-embedding algorithms for electronic structure calculations: Scaling up to complex molecular systems,”The Journal of Chemical Physics, vol. 154, no. 11, Mar. 2021. [Online]. Available: http://dx.doi.org/10.1063/5.0029536
-
[13]
Penalty methods for a variational quantum eigensolver,
K. Kuroiwa and Y . O. Nakagawa, “Penalty methods for a variational quantum eigensolver,”Phys. Rev. Res., vol. 3, p. 013197, Feb 2021. [Online]. Available: https://link.aps.org/doi/10.1103/PhysRevResearch.3.013197
-
[14]
Homo–lumo gaps of homogeneous polycyclic aromatic hydrocarbon clusters,
D. Chen and H. Wang, “Homo–lumo gaps of homogeneous polycyclic aromatic hydrocarbon clusters,”The Journal of Physical Chemistry C, vol. 123, no. 45, pp. 27 785–27 793, 2019. [Online]. Available: https://doi.org/10.1021/acs.jpcc.9b08300
-
[15]
Pyscf: the python-based simulations of chemistry framework,
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, “Pyscf: the python-based simulations of chemistry framework,”WIREs Computational Molecular Science, vol. 8, no. 1, p. e1340, 2018. [Online]. Available: https://wires.onlinelibrary.wiley.com/doi/abs/10.1002...
-
[16]
Qiskit nature: A library for quantum simulation of chemistry and materials science,
Q. N. Developers, “Qiskit nature: A library for quantum simulation of chemistry and materials science,”arXiv preprint, 2024. [Online]. Available: https://qiskit.org/nature
2024
-
[17]
Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,
J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love, and A. Aspuru-Guzik, “Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,”Quantum Science and Technology, vol. 4, no. 1, p. 014008, oct 2018. [Online]. Available: https://doi.org/10.1088/2058-9565/aad3e4
-
[18]
E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y . Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff et al., “Software for the frontiers of quantum chemistry: An overview of developments in the q-chem 5 package,”The Journal of Chemical Physics, vol. 155, no. 8, p. 084801, 2021. [Online]. Available: https://doi.org/10.1063/5.0055522
-
[19]
Convergence acceleration of iterative sequences. the case of scf iteration,
P. Pulay, “Convergence acceleration of iterative sequences. the case of scf iteration,”Chemical Physics Letters, vol. 73, no. 2, pp. 393–398, 1980. [Online]. Available: https://www.sciencedirect.com/science/article/pii/0009261480803964
-
[20]
Tapering off qubits to simulate fermionic hamiltonians,
S. Bravyi, J. M. Gambetta, A. Mezzacapo, and K. Temme, “Tapering off qubits to simulate fermionic hamiltonians,”arXiv preprint,
-
[21]
Tapering off qubits to simulate fermionic Hamiltonians
[Online]. Available: https://arxiv.org/abs/1701.08213 June 30, 2026 DRAFT 17
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[22]
A limited memory algorithm for bound constrained optimization,
R. H. Byrd, P. Lu, J. Nocedal, and C. Zhu, “A limited memory algorithm for bound constrained optimization,”SIAM Journal on Scientific Computing, vol. 16, no. 5, pp. 1190–1208, 1995. [Online]. Available: https://doi.org/10.1137/0916069
-
[23]
Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons,
M. V . Roux, M. Temprado, J. S. Chickos, and Y . Nagano, “Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons,”Journal of Physical and Chemical Reference Data, vol. 37, no. 4, pp. 1855–1996, 2008. [Online]. Available: https://pubs.aip.org/aip/jpr/article-abstract/37/4/1855/912308/Critically-Evaluated-Thermochemical-Properties-of
1996
-
[24]
Calculation of vibrationally resolved absorption spectra of acenes and pyrene,
I. Benkyi, E. Tapavicza, H. Fliegl, and D. Sundholm, “Calculation of vibrationally resolved absorption spectra of acenes and pyrene,” Phys. Chem. Chem. Phys., vol. 21, pp. 21 094–21 103, 2019. [Online]. Available: http://dx.doi.org/10.1039/C9CP04178H
-
[25]
Rotational analysis of the 2600a absorption system of benzene,
J. H. Callomom, T. M. Dunn, and I. M. Mills, “Rotational analysis of the 2600a absorption system of benzene,”Philosophical Transactions of the Royal Society of London, Series A: Mathematical and Physical Sciences, vol. 259, no. 1104, pp. 499–532, 05 1966. [Online]. Available: https://doi.org/10.1098/rsta.1966.0023
-
[26]
N. O. C. Winter, N. K. Graf, S. Leutwyler, and C. H ¨attig, “Benchmarks for 0–0 transitions of aromatic organic molecules: Dft/b3lyp, adc(2), cc2, sos-cc2 and scs-cc2 compared to high-resolution gas-phase data,”Phys. Chem. Chem. Phys., vol. 15, pp. 6623–6630, 2013. [Online]. Available: http://dx.doi.org/10.1039/C2CP42694C
-
[27]
Absorption spectroscopy of astrophysically relevant molecules in supersonic jets,
F. Huisken, G. Rouill ´e, Y . Carpentier, M. Steglich, and T. Henning, “Absorption spectroscopy of astrophysically relevant molecules in supersonic jets,”AIP Conference Proceedings, vol. 1333, no. 1, pp. 819–824, 05 2011. [Online]. Available: https://doi.org/10.1063/1.3562747
-
[28]
Experimental determination and interpretation of the fluorescence and fluorescence excitation spectra of chrysene cooled in a supersonic jet,
N. A. Borisevich, G. G. Dyachenko, V . A. Petukhov, and M. A. Semenov, “Experimental determination and interpretation of the fluorescence and fluorescence excitation spectra of chrysene cooled in a supersonic jet,”Optics and Spectroscopy, vol. 105, pp. 859–866,
-
[29]
Available: https://api.semanticscholar.org/CorpusID:120301445
[Online]. Available: https://api.semanticscholar.org/CorpusID:120301445
-
[30]
Charge transport in organic semiconductors,
V . Coropceanu, J. Cornil, D. A. da Silva Filho, Y . Olivier, R. Silbey, and J.-L. Br ´edas, “Charge transport in organic semiconductors,” Chemical Reviews, vol. 107, no. 4, pp. 926–952, 2007, pMID: 17378615. [Online]. Available: https://doi.org/10.1021/cr050140x
-
[31]
Density functional theory and the band gap problem,
J. P. Perdew, “Density functional theory and the band gap problem,”International Journal of Quantum Chemistry, vol. 28, no. S19, pp. 497–523, 1985. [Online]. Available: https://onlinelibrary.wiley.com/doi/abs/10.1002/qua.560280846
-
[32]
Scan: An efficient density functional yielding accurate structures and energies of diversely-bonded materials,
J. Sun, R. C. Remsing, Y . Zhang, Z. Sun, A. Ruzsinszky, H. Peng, Z. Yang, A. Paul, U. Waghmare, X. Wu, M. L. Klein, and J. P. Perdew, “Scan: An efficient density functional yielding accurate structures and energies of diversely-bonded materials,”arXiv preprint,
-
[33]
[Online]. Available: https://arxiv.org/abs/1511.01089
work page internal anchor Pith review Pith/arXiv arXiv
-
[34]
Tuned range-separated hybrids in density functional theory,
R. Baer, E. Livshits, and U. Salzner, “Tuned range-separated hybrids in density functional theory,”Annual Review of Physical Chemistry, vol. 61, no. V olume 61, 2010, pp. 85–109, 2010. [Online]. Available: https://www.annualreviews.org/content/journals/10.1146/annurev. physchem.012809.103321
-
[35]
A variational eigenvalue solver on a photonic quantum processor,
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’Brien, “A variational eigenvalue solver on a photonic quantum processor,”Nature Communications, vol. 5, no. 1, 2014. [Online]. Available: http://dx.doi.org/10.1038/ncomms5213
-
[36]
Quantum computing in the nisq era and beyond,
J. Preskill, “Quantum computing in the nisq era and beyond,”Quantum, vol. 2, p. 79, Aug. 2018. [Online]. Available: http://dx.doi.org/10.22331/q-2018-08-06-79
work page internal anchor Pith review doi:10.22331/q-2018-08-06-79 2018
-
[37]
Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, “Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,”Nature, vol. 549, no. 7671, p. 242–246, 2017. [Online]. Available: http://dx.doi.org/10.1038/nature23879
-
[38]
Half-metallic graphene nanoribbons,
Y .-W. Son, M. L. Cohen, and S. G. Louie, “Half-metallic graphene nanoribbons,”Nature, vol. 444, no. 7117, pp. 347–349, 2006. [Online]. Available: https://www.nature.com/articles/nature05180 June 30, 2026 DRAFT
2006
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.