LDARNet: DNA Adaptive Representation Network with Learnable Tokenization for Genomic Modeling
Pith reviewed 2026-06-28 06:15 UTC · model grok-4.3
The pith
LDARNet shows that unsupervised adaptive token boundaries in a 120M genomic model drive state-of-the-art histone modification performance and align with known biological motifs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
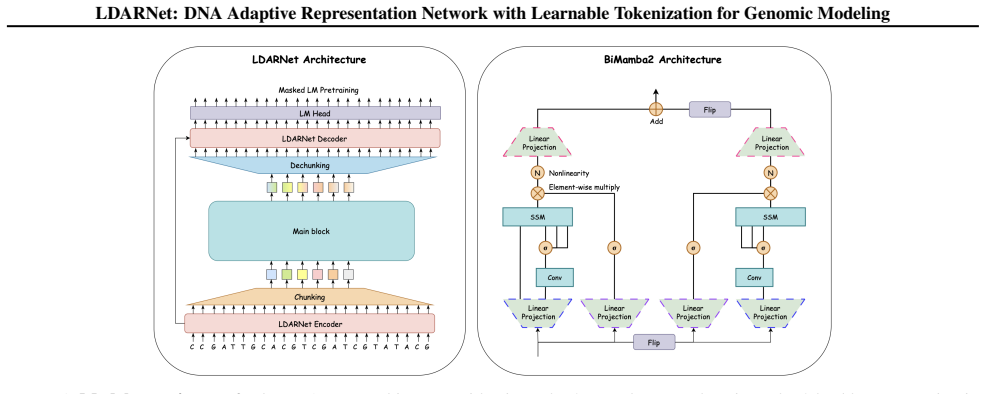
LDARNet is a hierarchical 120M-parameter genomic foundation model that adapts H-Net-style dynamic chunking to masked language modeling through BiMamba-2 layers, local attention, bidirectional routing, and a ratio-based regularizer, thereby inducing adaptive token boundaries. Fine-tuning on the Nucleotide Transformer and Genomic Benchmarks suites yields state-of-the-art results on five histone modification tasks while outperforming models up to 20 times larger; a matched-compute ablation confirms that learned boundaries outperform fixed-grid tokenization by up to 14 percentage points on those tasks. Nucleotide-level analysis shows the unsupervised boundaries align with promoter motifs and spl
What carries the argument
Bidirectional routing combined with a ratio-based regularizer that learns adaptive token boundaries during masked language modeling training.
If this is right
- Learned boundaries outperform fixed-grid tokenization by up to 14 percentage points on histone tasks at identical compute.
- The model records state-of-the-art results on five histone modification tasks and wins on 11 of 18 tasks among models under 300M parameters.
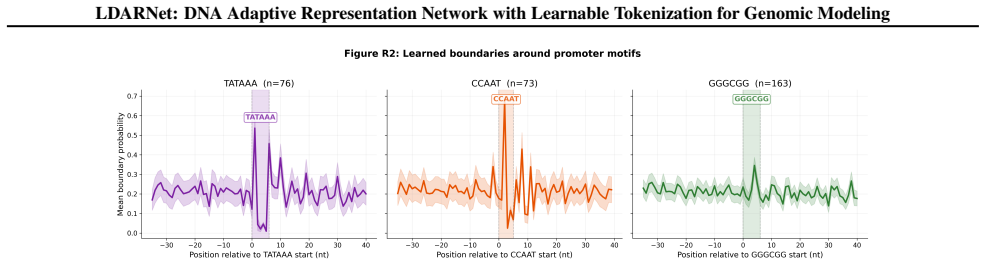
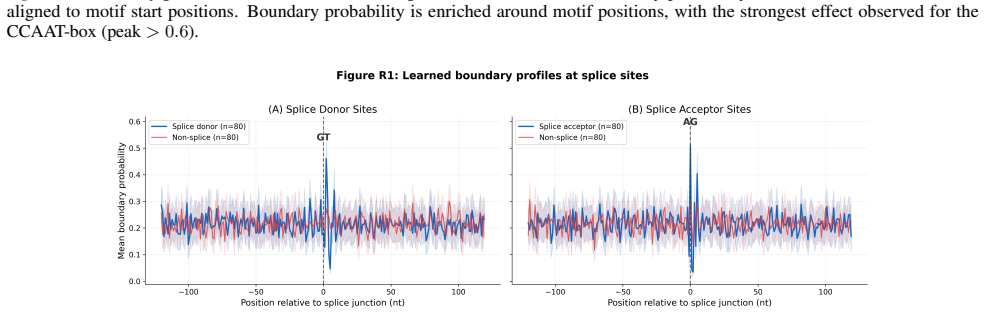
- Unsupervised boundaries align with canonical promoter motifs and splice junctions.
- The approach supplies a biological interpretation for why adaptive tokenization benefits genomic foundation models.
Where Pith is reading between the lines
- Similar routing mechanisms could be tested on protein sequences to check whether they discover functional domain boundaries.
- If the alignment with known motifs holds at larger scale, the method might surface previously unrecognized regulatory elements.
- The efficiency gains from adaptive chunking suggest potential applicability to long-context sequence tasks beyond genomics.
Load-bearing premise
The ratio-based regularizer and bidirectional routing produce token boundaries that are both biologically meaningful and causally responsible for the performance gains rather than other model components or training choices.
What would settle it
An ablation that disables adaptive routing, keeps compute and all other components identical, and shows no drop in histone task accuracy would falsify the claim that learned boundaries drive the reported gains.
Figures
read the original abstract
Genomic foundation models increasingly adopt large language model architectures, yet almost universally rely on fixed tokenization schemes such as $k$-mers, BPE, or single nucleotides, which impose arbitrary sequence boundaries that may obscure biologically relevant structure. We present LDARNet, a 120M-parameter hierarchical genomic foundation model that adapts H-Net-style dynamic chunking from autoregressive generation to masked language modeling, combining BiMamba-2 state-space layers with local attention, bidirectional routing, and a ratio-based regularizer to induce adaptive token boundaries without supervision. Fine-tuned on 27 tasks from the Nucleotide Transformer and Genomic Benchmarks suites, LDARNet achieves 11/18 wins among compact models ($<$300M parameters) and state-of-the-art results on 5 histone modification tasks, outperforming models up to 20$\times$ larger. A FLOPs-matched controlled experiment isolates learned routing as the source of these gains: learned boundaries beat fixed-grid boundaries by up to 14 percentage points on histone tasks at identical compute. Nucleotide-resolution analysis further shows that the learned boundaries align with canonical promoter motifs and splice junctions without supervision, providing a biological interpretation for adaptive tokenization in genomic foundation models.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. LDARNet is a 120M-parameter hierarchical genomic foundation model adapting H-Net-style dynamic chunking to masked language modeling. It combines BiMamba-2 state-space layers with local attention, bidirectional routing, and a ratio-based regularizer to induce unsupervised adaptive token boundaries. On 27 tasks from the Nucleotide Transformer and Genomic Benchmarks suites, the model reports 11/18 wins among compact models (<300M parameters) and state-of-the-art results on 5 histone modification tasks, outperforming models up to 20× larger. A FLOPs-matched controlled experiment claims learned boundaries outperform fixed-grid boundaries by up to 14 percentage points on histone tasks at identical compute, with nucleotide-resolution analysis showing alignment to canonical promoter motifs and splice junctions.
Significance. If the FLOPs-matched experiment correctly isolates the contribution of learned token boundaries, the work would demonstrate that adaptive tokenization can yield both performance gains and biologically interpretable structure in genomic foundation models. The controlled comparison and the reported alignment with known motifs are strengths that would advance the field beyond fixed k-mer or BPE schemes.
major comments (2)
- [§4.3] §4.3 (FLOPs-matched controlled experiment): The manuscript does not provide equations, pseudocode, or implementation details for the fixed-grid baseline. It is therefore impossible to verify that the baseline differs from the learned version solely in boundary selection while preserving identical BiMamba-2 routing logic, state-space updates, parameter count, and local attention masking. Without this isolation, the reported 14pp gap on histone tasks cannot be attributed to adaptive boundaries.
- [Results section] Results section, histone modification tasks: The SOTA claims and the 14pp improvement are presented without error bars, standard deviations across runs, or details on hyperparameter search procedures. This weakens the claim that the gains are robustly due to the ratio-based regularizer and bidirectional routing rather than training details.
minor comments (2)
- [Abstract / Table 1] The abstract states '11/18 wins among compact models' but the corresponding table would benefit from explicit column headers indicating which models are included in the 18 and whether ties are counted.
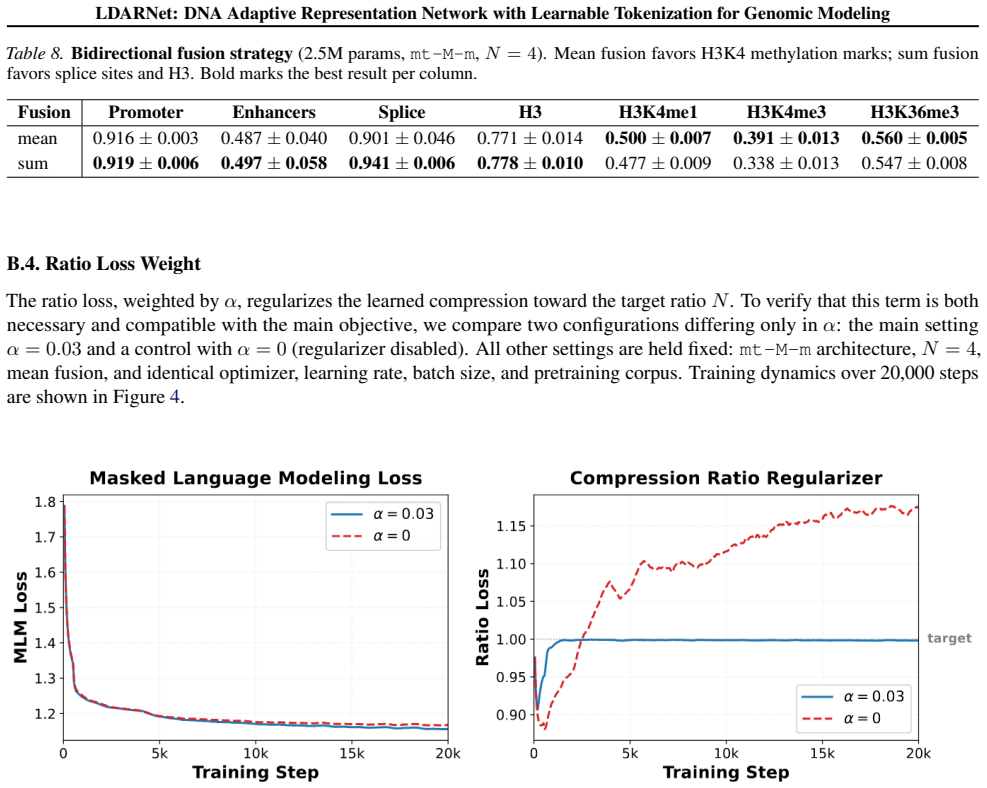
- [§3.2] Notation for the ratio-based regularizer is introduced without a dedicated equation number; cross-referencing to the loss term in §3.2 would improve clarity.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on LDARNet. We address each major comment below and commit to revisions that strengthen the manuscript without altering its core claims.
read point-by-point responses
-
Referee: [§4.3] §4.3 (FLOPs-matched controlled experiment): The manuscript does not provide equations, pseudocode, or implementation details for the fixed-grid baseline. It is therefore impossible to verify that the baseline differs from the learned version solely in boundary selection while preserving identical BiMamba-2 routing logic, state-space updates, parameter count, and local attention masking. Without this isolation, the reported 14pp gap on histone tasks cannot be attributed to adaptive boundaries.
Authors: We agree that the current version of §4.3 lacks the requested implementation details. In the revision we will insert the precise equations for the fixed-grid boundary generation, pseudocode for the controlled experiment, and explicit statements confirming that the baseline reuses identical BiMamba-2 routing logic, state-space updates, parameter count, and local attention masking, differing only in boundary selection. This addition will make the isolation verifiable and directly support attribution of the observed gap to adaptive tokenization. revision: yes
-
Referee: Results section, histone modification tasks: The SOTA claims and the 14pp improvement are presented without error bars, standard deviations across runs, or details on hyperparameter search procedures. This weakens the claim that the gains are robustly due to the ratio-based regularizer and bidirectional routing rather than training details.
Authors: We acknowledge that the absence of error bars and hyperparameter details in the current results section limits the strength of the robustness argument. The revised manuscript will report mean performance and standard deviation across at least three independent random seeds for all histone modification tasks, together with a concise description of the hyperparameter search protocol (grid ranges, selection criterion, and final values). These additions will allow readers to assess whether the reported gains are attributable to the architectural components rather than training variance. revision: yes
Circularity Check
No circularity: empirical claims rest on controlled experiments without reduction to fitted inputs or self-citations
full rationale
The paper's central claims (SOTA on histone tasks, 14pp gap from learned vs fixed boundaries) are presented as outcomes of fine-tuning on external benchmarks and a FLOPs-matched ablation. The provided abstract and reader summary contain no equations, parameter-fitting procedures, or self-citations that would make the reported performance gains equivalent to the inputs by construction. No load-bearing step matches any of the enumerated circularity patterns; the derivation chain is self-contained against the described experimental controls.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URL https://www.biorxiv.org/content/ early/2021/04/08/2021.04.07.438649
doi: 10.1101/2021.04.07.438649. URL https://www.biorxiv.org/content/ early/2021/04/08/2021.04.07.438649. Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J., Lopez Carranza, N., Grzywaczewski, A. H., Oteri, F., Dallago, C., Trop, E., de Almeida, B. P., Sirelkhatim, H., et al. Nucleotide transformer: building and evaluating robust foundation models for huma...
-
[2]
Dao, T. and Gu, A. Transformers are ssms: Generalized models and efficient algorithms through structured state space duality.arXiv preprint arXiv:2405.21060,
work page internal anchor Pith review Pith/arXiv arXiv
-
[3]
Janusdna: A powerful bi-directional hybrid dna foundation model.arXiv preprint arXiv:2505.17257,
Duan, Q., Huang, B., Song, Z., Lehmann, I., Gu, L., Eils, R., and Wild, B. Janusdna: A powerful bi-directional hybrid dna foundation model.arXiv preprint arXiv:2505.17257,
-
[4]
arXiv preprint arXiv:2507.07955 , year=
Hwang, S., Wang, B., and Gu, A. Dynamic chunking for end-to-end hierarchical sequence modeling.arXiv preprint arXiv:2507.07955,
- [5]
-
[6]
Decoupled Weight Decay Regularization
Loshchilov, I. and Hutter, F. Decoupled weight decay regu- larization.arXiv preprint arXiv:1711.05101,
work page internal anchor Pith review Pith/arXiv arXiv
-
[7]
Carbon Emissions and Large Neural Network Training
URL https://arxiv.org/abs/2104.10350. Qiao, L., Ye, P., Ren, Y ., Bai, W., Liang, C., Ma, X., Dong, N., and Ouyang, W. Model decides how to tokenize: Adaptive dna sequence tokenization with mxdna.Ad- vances in Neural Information Processing Systems, 37: 66080–66107,
work page internal anchor Pith review Pith/arXiv arXiv
-
[8]
Schiff, Y ., Kao, C.-H., Gokaslan, A., Dao, T., Gu, A., and Kuleshov, V
doi: 10.1038/s42256-024-00872-0. Schiff, Y ., Kao, C.-H., Gokaslan, A., Dao, T., Gu, A., and Kuleshov, V . Caduceus: Bi-directional equivariant long- range dna sequence modeling.Proceedings of machine learning research, 235:43632,
-
[9]
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A
URL https: //arxiv.org/abs/2404.14408. Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser, L., and Polosukhin, I. Attention is all you need.CoRR, abs/1706.03762,
-
[10]
URL http://arxiv.org/abs/1706.03762. Wang, J., Gangavarapu, T., Yan, J. N., and Rush, A. M. Mambabyte: Token-free selective state space model,
work page internal anchor Pith review Pith/arXiv arXiv
-
[11]
Wu, W., Li, Q., Li, M., Fu, K., Feng, F., Ye, J., Xiong, H., and Wang, Z
URL https://arxiv.org/abs/ 2401.13660. Wu, W., Li, Q., Li, M., Fu, K., Feng, F., Ye, J., Xiong, H., and Wang, Z. Generator: A long-context genera- tive genomic foundation model,
-
[12]
Zhou, Z., Ji, Y ., Li, W., Dutta, P., Davuluri, R., and Liu, H
URL https: //arxiv.org/abs/2502.07272. Zhou, Z., Ji, Y ., Li, W., Dutta, P., Davuluri, R., and Liu, H. Dnabert-2: Efficient foundation model and benchmark for multi-species genome.arXiv preprint arXiv:2306.15006,
-
[13]
Gradients are clipped to a maximum norm of 1.0
using β1 = 0.9 , β2 = 0.95 (following Dao & Gu (2024)), ϵ= 10 −8, and weight decay 0.01. Gradients are clipped to a maximum norm of 1.0. The learning rate follows a warmup–stable–decay (WSD) schedule with base rate 5×10 −4: linear warmup over the first 10% of training, a constant plateau for70%, and inverse-square-root decay over the final20%ending at5%of...
2024
-
[14]
is a Transformer-based model trained in a supervised multi-task fashion to predict thousands of chromatin and gene expression tracks (from ENCODE and Roadmap Epigenomics) directly from DNA sequence. While the other compact baselines use self-supervised pretraining objectives (MLM or next-token prediction), Enformer is used here as a frozen-torso pretraine...
2025
-
[15]
Training used sequences of length 4096 with effective batch size 512 and required 7 days of wall-clock time, totaling1,008 GPU-hours (42 GPU-days)
(∼300 B base pairs). Training used sequences of length 4096 with effective batch size 512 and required 7 days of wall-clock time, totaling1,008 GPU-hours (42 GPU-days). Downstream Evaluation.For each task, we performed an exhaustive hyperparameter search over 36 configurations (9 learning rates × 4 batch sizes), followed by 10-fold cross-validation with t...
2021
-
[16]
Code and pretrained model weights will be made available at https://github.com/darlednik/ ICML-LDARNet
and the Genomic Benchmarks suite (Greˇsov´a et al., 2023). Code and pretrained model weights will be made available at https://github.com/darlednik/ ICML-LDARNet. 19
2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.