The effect of normal stress on stacking fault energy in face-centered cubic metals
Pith reviewed 2026-05-16 16:48 UTC · model grok-4.3
The pith
Normal compression perpendicular to (111) planes raises both stable and unstable stacking fault energies in FCC metals while tension lowers them.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
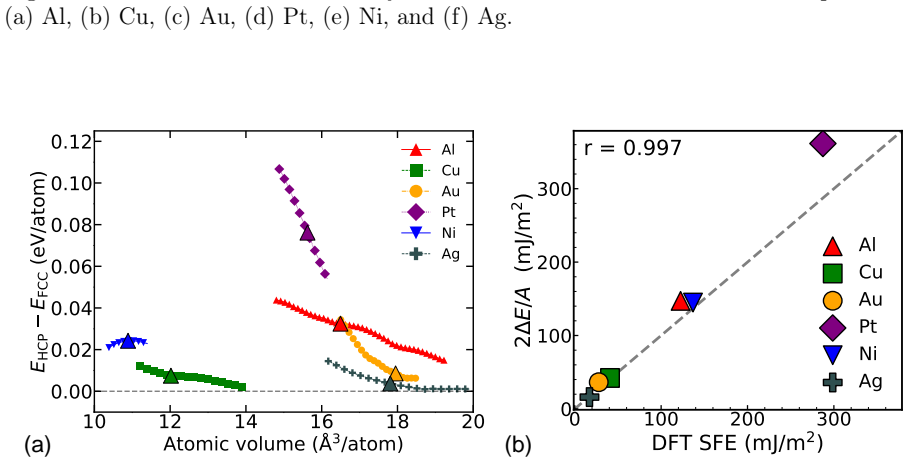
Density functional theory calculations on Al, Ni, Cu, Ag, Au, and Pt demonstrate that normal compression increases both stable and unstable stacking fault energies on (111) planes while normal tension decreases them. Stacking fault formation is accompanied by an inelastic expansion normal to the plane. Many interatomic potentials fail to reproduce these trends, often predicting the opposite effect.
What carries the argument
DFT supercell calculations of stacking fault energy under applied normal stress, incorporating the observed inelastic normal-direction expansion during fault formation.
If this is right
- Dislocation cross-slip rates and energies will vary with local normal stress in ways not captured by zero-stress models.
- Dislocation nucleation at interfaces and crack tips will be suppressed under compression and facilitated under tension.
- Classical and machine-learning potentials must be retrained or reformulated to reproduce the normal-stress effect on fault energies.
- High-pressure deformation and fracture simulations require explicit inclusion of this stress dependence.
Where Pith is reading between the lines
- The inelastic expansion during fault formation may couple to hydrostatic pressure effects in polycrystalline samples.
- Training data for machine-learning potentials should include stressed fault configurations to avoid the failures seen in current potentials.
- This stress dependence could alter predictions of twinning versus slip competition under extreme loading.
Load-bearing premise
The chosen DFT exchange-correlation functional, k-point sampling, and supercell size correctly capture the normal-direction inelastic response and its stress dependence without systematic bias.
What would settle it
A direct experimental measurement of stacking fault energy under controlled normal compression in one of the six metals that shows a decrease rather than an increase would falsify the central DFT result.
Figures
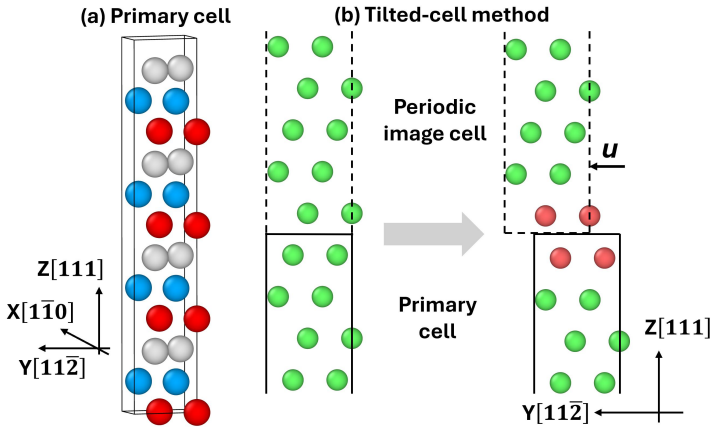




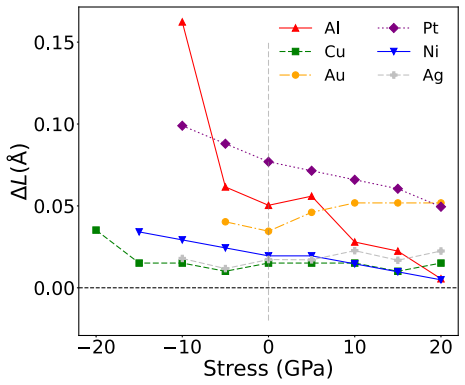





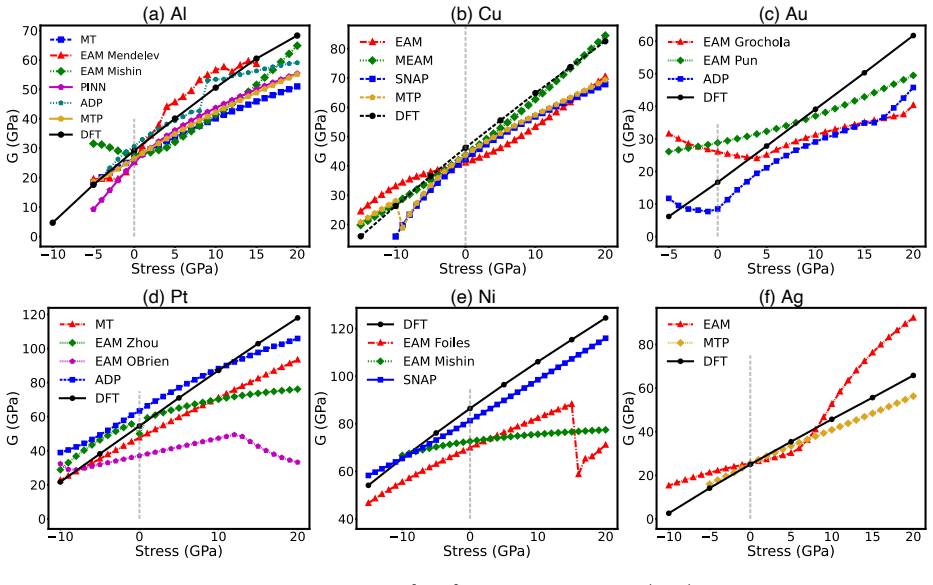
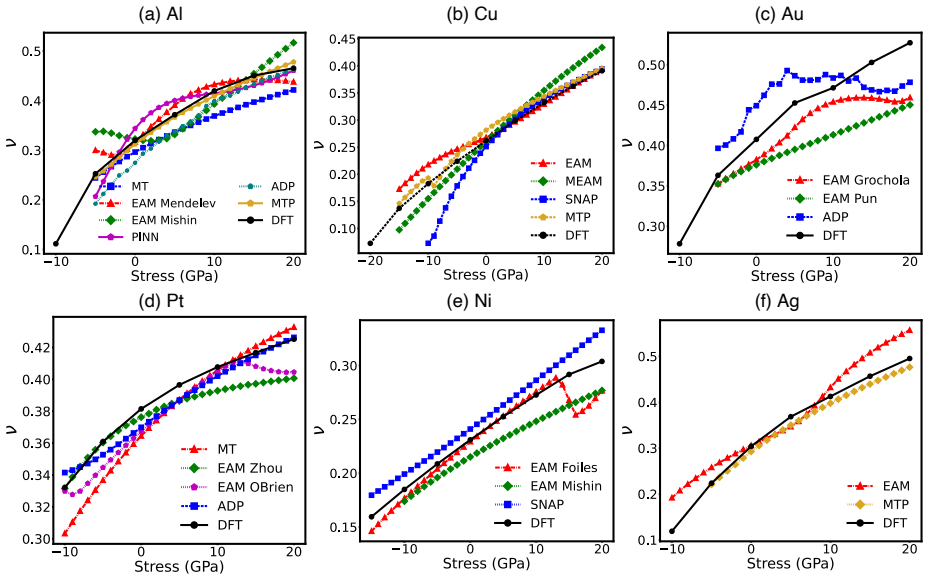




read the original abstract
Plastic deformation and fracture of FCC metals involve the formation of stable or unstable stacking faults (SFs) on (111) plane. Examples include dislocation cross-slip and dislocation nucleation at interfaces and near crack tips. The stress component normal to (111) plane can strongly affect the SF energy when the stress magnitude reaches several to tens of GPa. We conduct a series of DFT calculations of SF energies in six FCC metals: Al, Ni, Cu, Ag, Au, and Pt. The results show that normal compression significantly increases the stable and unstable SF energies in all six metals, while normal tension decreases them. The SF formation is accompanied by inelastic expansion in the normal direction. The DFT calculations are compared with predictions of several representative classical and machine-learning interatomic potentials. Many potentials fail to capture the correct stress effect on the SF energy, often predicting trends opposite to the DFT calculations. Possible ways to improve the ability of potentials to represent the stress effect on SF energy are discussed.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses density functional theory (DFT) to compute the effect of normal stress on stable and unstable stacking fault (SF) energies in six FCC metals (Al, Ni, Cu, Ag, Au, Pt). It reports that normal compression increases both energies while tension decreases them, and that SF formation produces inelastic expansion normal to the (111) plane. DFT results are compared against multiple classical and machine-learning interatomic potentials, many of which predict incorrect or opposite stress trends; possible improvements to the potentials are discussed.
Significance. If the central DFT trends hold, the work provides a direct numerical benchmark for stress-dependent SF energies at GPa-level normal stresses relevant to fracture and dislocation nucleation. The explicit comparison to interatomic potentials and discussion of their deficiencies is a strength, as it supplies concrete guidance for model improvement in high-stress regimes.
major comments (2)
- [Methods] Methods section: explicit convergence tests with respect to supercell thickness (number of (111) layers) and k-point sampling are not shown under the applied normal stresses (several to tens of GPa). Because the inelastic normal expansion and the derivative of SF energy with respect to normal stress both depend on accurate out-of-plane relaxation, periodic-image coupling in small supercells could systematically bias the reported expansion and stress dependence.
- [Results] Results, inelastic-expansion paragraph: the quantitative expansion values are given without error bars or sensitivity checks to computational parameters. This leaves the magnitude of the inelastic response, which is load-bearing for the central claim, without a clear uncertainty estimate.
minor comments (3)
- [Abstract] Abstract: the range of normal stresses examined should be stated explicitly to contextualize the reported trends.
- [Comparison with potentials] Comparison section: a summary table listing each potential and whether it reproduces, reverses, or fails to capture the DFT stress dependence would improve clarity.
- [Figures] Figure captions: include the exact normal-stress values used in each panel for immediate reference.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which help improve the clarity and robustness of our manuscript. We address each major comment below and have revised the manuscript to incorporate additional convergence data and uncertainty estimates.
read point-by-point responses
-
Referee: [Methods] Methods section: explicit convergence tests with respect to supercell thickness (number of (111) layers) and k-point sampling are not shown under the applied normal stresses (several to tens of GPa). Because the inelastic normal expansion and the derivative of SF energy with respect to normal stress both depend on accurate out-of-plane relaxation, periodic-image coupling in small supercells could systematically bias the reported expansion and stress dependence.
Authors: We agree that explicit convergence tests under applied normal stress are necessary. Our original calculations employed a 12-layer (111) supercell with a 12×12×1 Γ-centered k-point mesh, which had been converged at zero stress. To address the referee's concern, we have performed additional tests under normal stresses from −10 GPa to +10 GPa using supercells with 8, 12, and 18 layers and k-point meshes up to 18×18×1. The stable and unstable SF energies change by less than 2 meV/Ų and the inelastic normal expansion by less than 0.005 Å when the supercell thickness is increased from 12 to 18 layers. These results confirm that periodic-image coupling does not materially affect the reported trends. We will add a dedicated convergence subsection and supporting table to the Methods section in the revised manuscript. revision: yes
-
Referee: [Results] Results, inelastic-expansion paragraph: the quantitative expansion values are given without error bars or sensitivity checks to computational parameters. This leaves the magnitude of the inelastic response, which is load-bearing for the central claim, without a clear uncertainty estimate.
Authors: We acknowledge that the inelastic-expansion values were presented without explicit uncertainty estimates. We have now carried out sensitivity analyses by varying the plane-wave cutoff energy (400–600 eV) and k-point density while keeping the supercell fixed. Across all six metals the inelastic normal expansion varies by at most 0.01 Å, which we adopt as the uncertainty. We will report these values with error bars in the revised Results section and add a short paragraph discussing the computational sensitivity of the inelastic response. revision: yes
Circularity Check
No circularity: central results are direct DFT outputs
full rationale
The paper performs a series of DFT calculations to obtain stacking fault energies under applied normal stress for six FCC metals. The reported trends (compression increases stable/unstable SF energies, tension decreases them, accompanied by inelastic normal expansion) are numerical results from these computations, not obtained by fitting parameters to a subset of data and then predicting related quantities, nor by self-referential equations or self-citation chains that reduce the claims to inputs by construction. Comparisons to classical and ML potentials serve as external benchmarks rather than load-bearing justifications. No load-bearing step matches any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard DFT exchange-correlation functionals and pseudopotentials sufficiently capture the electronic response to normal stress on (111) planes in FCC metals.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We conduct a series of DFT calculations of SF energies in six FCC metals... normal compression significantly increases the stable and unstable SF energies... The SF formation is accompanied by inelastic expansion in the normal direction.
-
IndisputableMonolith/Foundation/AlphaCoordinateFixation.leanJ_uniquely_calibrated_via_higher_derivative unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The DFT calculations are compared with predictions of several representative classical and machine-learning interatomic potentials.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Forward citations
Cited by 1 Pith paper
-
Interatomic potentials for platinum
Two new interatomic potentials for platinum trained only on DFT data outperform existing models in matching quantum calculations and experimental properties.
Reference graph
Works this paper leans on
-
[1]
P. M. Anderson, J. P. Hirth, J. Lothe, Theory of dislocations, Cambridge University Press, 2017
work page 2017
-
[2]
J. A. Zimmerman, H. Gao, F. F. Abraham, Generalized stacking fault energies for embedded atom fcc metals, Modelling and Simulation in Materials Science and Engi- neering 8 (2000) 103
work page 2000
-
[3]
G. Lu, N. Kioussis, V. V. Bulatov, E. Kaxiras, Generalized-stacking-fault energy surface and dislocation properties of aluminum, Phys. Rev. B 62 (2000) 3099–3108
work page 2000
-
[4]
J. A. Yan, C. Y. Wang, S. Y. Wang, Generalized stacking fault energy and dislocation properties in bcc Fe: A first-principles study, Phys. Rev. B 70 (2004) 174105
work page 2004
-
[5]
C. Wang, H. Wang, T. Huang, X. Xue, F. Que, Q. Jiang, Generalized-stacking-fault energy and twin-boundary energy of hexagonal close-packed Au: A first-principles calculation, Scientific Reports 5 (2015) 10213
work page 2015
-
[6]
V. Bulatov, F. F. Abraham, L. Kubin, B. Devincre, S. Yip, Connecting atomistic and mesoscale simulations of crystal plasticity, Nature 391 (1998) 669–672
work page 1998
-
[7]
J. R. Rice, Dislocation nucleation from a crack tip: an analysis based on the peierls concept, J. Mech. Phys. Solids 40 (1992) 239–271
work page 1992
-
[8]
G. S. Liu, X. Cheng, J. Wang, K. G. Chen, Y. Shen, Improvement of nonlocal Peierls- Nabarro models, Comp. Mater. Sci. 131 (2017) 69–77
work page 2017
-
[9]
G. Lu, N. Kioussis, V. V. Bulatov, E. Kaxiras, The Peierls-Nabarro model revisited, Philos. Mag. Lett. 80 (2000) 675–682
work page 2000
-
[10]
Schoeck, The core structure of dislocations
G. Schoeck, The core structure of dislocations. Peierls model vs. atomic simulations in Pd, Compu. Mater. Sci. 21 (2001) 124–134
work page 2001
-
[11]
G. Lu, The Peierls—Nabarro model of dislocations: A venerable theory and its current development, Springer Netherlands, Dordrecht, 2005, pp. 793–811
work page 2005
-
[12]
I. Beyerlein, A. Hunter, Understanding dislocation mechanics at the mesoscale using phase field dislocation dynamics, Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 374 (2016) 20150166
work page 2016
-
[13]
H. Kim, N. Mathew, D. J. Luscher, A. Hunter, Phase field dislocation dynamics (PFDD) modeling of non-Schmid behavior in BCC metals informed by atomistic sim- ulations, Journal of the Mechanics and Physics of Solids 152 (2021) 104460. 16
work page 2021
-
[14]
A. Hunter, I. J. Beyerlein, T. C. Germann, M. Koslowski, Influence of the stacking fault energy surface on partial dislocations in fcc metals with a three-dimensional phase field dislocations dynamics model, Physical Review B—Condensed Matter and Materials Physics 84 (2011) 144108
work page 2011
-
[15]
V. Borovikov, M. I. Mendelev, A. H. King, Effects of stable and unstable stacking fault energy on dislocation nucleation in nano-crystalline metals, Modelling and Simulation in Materials Science and Engineering 24 (2016) 085017
work page 2016
-
[16]
P. M. Anderson, J. R. Rice, Dislocation emission from cracks in crystals or along crystal interfaces, Scr Metall 20 (1986) 1467–1472
work page 1986
-
[17]
G. Xu, C. Zhang, Analysis of dislocation nucleation from a crystal surface based on the Peierls–Nabarro dislocation model, Journal of the Mechanics and Physics of Solids 51 (2003) 1371–1394
work page 2003
-
[18]
R. Cao, C. Deng, The ultra-small strongest grain size in nanocrystalline Ni nanowires, Scripta Mater. 94 (2015) 9–12
work page 2015
-
[19]
Q.-J. Li, B. Xu, S. Hara, J. Li, E. Ma, Sample-size-dependent surface dislocation nucleation in nanoscale crystals, Acta Materialia 145 (2018) 19–29
work page 2018
-
[20]
D. Mordehai, O. David, R. Kositski, Nucleation-controlled plasticity of metallic nanowires and nanoparticles, Advanced Materials 30 (2018) 1706710
work page 2018
-
[21]
J.Amodeo, K.Lizoul, Mechanicalpropertiesanddislocationnucleationinnanocrystals with blunt edges, Materials & Design 135 (2017) 223–231
work page 2017
- [22]
- [23]
-
[24]
J. Yan, Z. Zhang, H. Yu, K. Li, Q. Hu, J. Yang, Z. Zhang, Effects of pressure on the generalized stacking fault energy and twinning propensity of face-centered cubic metals, Journal of Alloys and Compounds 866 (2021) 158869
work page 2021
- [25]
- [26]
-
[27]
P. Branicio, J. Zhang, D. Srolovitz, Effect of strain on the stacking fault energy of copper: a first-principles study, Physical Review B—Condensed Matter and Materials Physics 88 (2013) 064104. 17
work page 2013
- [28]
-
[29]
M. Tschopp, D. McDowell, Influence of single crystal orientation on homogeneous dislocation nucleation under uniaxial loading, Journal of the Mechanics and Physics of Solids 56 (2008) 1806–1830
work page 2008
- [30]
-
[31]
Mishin, Machine-learning interatomic potentials for materials science, Acta Mater
Y. Mishin, Machine-learning interatomic potentials for materials science, Acta Mater. 214 (2021) 116980
work page 2021
-
[32]
Y.-W. Zhang, V. Sorkin, Z. H. Aitken, A. Politano, J. Behler, A. P Thompson, T. W. Ko, S. P. Ong, O. Chalykh, D. Korogod, E. Podryabinkin, A. Shapeev, J. Li, Y. Mishin, Z. Pei, X. Liu, J. Kim, Y. Park, S. Hwang, S. Han, K. Sheriff, Y. Cao, R. Freitas, Roadmap for the development of machine learning-based interatomic potentials, Mod- elling and Simulation ...
work page 2025
-
[33]
M. Jhon, A. Glaeser, D. Chrzan, Computational study of stacking faults in sapphire using total energy methods, Physical Review B—Condensed Matter and Materials Physics 71 (2005) 214101
work page 2005
- [34]
- [35]
- [36]
-
[37]
P. E. Blöchl, Projector augmented-wave method, Physical review B 50 (1994) 17953
work page 1994
-
[38]
J. P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple, Physical review letters 77 (1996) 3865
work page 1996
-
[39]
J. F. Nye, Physical Properties of Crystals, Clarendon Press, Oxford, 1985
work page 1985
-
[40]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. In’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, et al., Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer physics communications 271 (2022) 108171
work page 2022
-
[41]
M. S. Daw, S. M. Foiles, M. I. Baskes, The embedded-atom method: a review of theory and applications, Materials Science Reports 9 (1993) 251–310. 18
work page 1993
-
[42]
M. I. Baskes, Modified embedded-atom potentials for cubic metals and impurities, Phys. Rev. B 46 (1992) 2727–2742
work page 1992
-
[43]
Y.Mishin, M.J.Mehl, D.A.Papaconstantopoulos, PhasestabilityintheFe-Nisystem: Investigation by first-principles calculations and atomistic simulations, Acta Mater. 53 (2005) 4029–4041
work page 2005
-
[44]
R. K. Koju, Y. Li, Y. Mishin, Comparison of interatomic potentials for platinum, In preparation, 2005
work page 2005
-
[45]
T. Kumagai, S. Izumi, S. Hara, S. Sakai, Development of bond-order potentials that can reproduce the elastic constants and melting point of silicon for classical molecular dynamics simulation, Comp. Mater. Sci. 39 (2007) 457–464
work page 2007
-
[46]
G. P. Purja Pun, Y. Mishin, Optimized interatomic potential for silicon and its appli- cation to thermal stability of silicene, Phys. Rev. B 95 (2017) 224103
work page 2017
-
[47]
G. P. Pun, V. Yamakov, J. Hickman, E. Glaessgen, Y. Mishin, Development of a general-purpose machine-learning interatomic potential for aluminum by the physically informed neural network method, Physical Review Materials 4 (2020) 113807
work page 2020
-
[48]
X.-G. Li, C. Hu, C. Chen, Z. Deng, J. Luo, S. P. Ong, Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals, Physical Review B 98 (2018) 094104
work page 2018
- [49]
-
[50]
G. S. Fanourgakis, V. Pontikis, G. Zérah, Phase stability and intrinsic stacking faults in aluminum under pressure, Physical Review B 67 (2003) 094102
work page 2003
-
[51]
L. M. Hale, Z. T. Trautt, C. A. Becker, Atomistic manipulation toolkit (atomman), National Institute of Standards and Technology, 2023
work page 2023
- [52]
-
[53]
M. Mendelev, M. Kramer, C. A. Becker, M. Asta, Analysis of semi-empirical in- teratomic potentials appropriate for simulation of crystalline and liquid Al and Cu, Philosophical Magazine 88 (2008) 1723–1750
work page 2008
- [54]
-
[55]
S. Starikov, I. Gordeev, Y. Lysogorskiy, L. Kolotova, S. Makarov, Optimized in- teratomic potential for study of structure and phase transitions in Si-Au and Si-Al systems, Computational Materials Science 184 (2020) 109891. 19
work page 2020
-
[56]
S.A.Etesami, E.Asadi, Moleculardynamicsfornearmeltingtemperaturessimulations of metals using modified embedded-atom method, Journal of Physics and Chemistry of Solids 112 (2018) 61–72
work page 2018
-
[57]
G. Grochola, S. P. Russo, I. K. Snook, On fitting a gold embedded atom method potential using the force matching method, The Journal of chemical physics 123 (2005)
work page 2005
-
[58]
G. P. Purja Pun, Au_GLJ10_3.eam.alloy (unpublished), NIST In- teratomic Potentials Repository, 2017–Purja-Pun-G-P–Au, 2017. URL: https://www.ctcms.nist.gov/potentials/system/Au/
work page 2017
-
[59]
X. W. Zhou, R. Johnson, H. N. Wadley, Misfit-energy-increasing dislocations in vapor- deposited CoFe/NiFe multilayers, Physical Review B 69 (2004) 144113
work page 2004
-
[60]
C. O’Brien, C. Barr, P. Price, K. Hattar, S. Foiles, Grain boundary phase transforma- tions in PtAu and relevance to thermal stabilization of bulk nanocrystalline metals, Journal of Materials Science 53 (2018) 2911–2927
work page 2018
-
[61]
S. M. Foiles, J. Hoyt, Computation of grain boundary stiffness and mobility from boundary fluctuations, Acta Materialia 54 (2006) 3351–3357
work page 2006
-
[62]
Y. Mishin, Atomistic modeling of theγandγ ′-phases of the Ni-Al system, Acta Materialia 52 (2004) 1451–1467
work page 2004
-
[63]
Y. Zuo, C. Chen, X. Li, Z. Deng, Y. Chen, J. Behler, G. Csányi, A. V. Shapeev, A. P. Thompson, M. A. Wood, et al., Performance and cost assessment of machine learning interatomic potentials, The Journal of Physical Chemistry A 124 (2020) 731–745
work page 2020
-
[64]
P. Williams, Y. Mishin, J. Hamilton, An embedded-atom potential for the Cu-Ag system, Modelling and Simulation in Materials Science and Engineering 14 (2006) 817
work page 2006
-
[65]
A. V. Shapeev, Moment tensor potentials: A class of systematically improvable inter- atomic potentials, Multiscale Modeling & Simulation 14 (2016) 1153–1173
work page 2016
- [66]
-
[67]
Vocadlo, et al., The ab initio melting curve of aluminium, arXiv preprint cond- mat/0108460 (2001)
L. Vocadlo, et al., The ab initio melting curve of aluminium, arXiv preprint cond- mat/0108460 (2001)
-
[68]
M. De Jong, W. Chen, T. Angsten, A. Jain, R. Notestine, A. Gamst, M. Sluiter, C. Krishna Ande, S. Van Der Zwaag, J. J. Plata, et al., Charting the complete elastic properties of inorganic crystalline compounds, Scientific data 2 (2015) 1–13
work page 2015
-
[69]
R. Tran, Z. Xu, B. Radhakrishnan, D. Winston, W. Sun, K. A. Persson, S. P. Ong, Surface energies of elemental crystals, Scientific data 3 (2016) 1–13. 20
work page 2016
-
[70]
R. Qiu, H. Lu, B. Ao, L. Huang, T. Tang, P. Chen, Energetics of intrinsic point defects in aluminium via orbital-free density functional theory, Philosophical Magazine 97 (2017) 2164–2181
work page 2017
- [71]
-
[72]
M. Iyer, V. Gavini, T. M. Pollock, Energetics and nucleation of point defects in aluminum under extreme tensile hydrostatic stresses, Physical Review B 89 (2014) 014108
work page 2014
-
[73]
T.Sjostrom, S.Crockett, S.Rudin, Multiphasealuminumequationsofstateviadensity functional theory, Physical Review B 94 (2016) 144101
work page 2016
-
[74]
J. F. Devlin, Stacking fault energies of be, mg, al, cu, ag, and au, Journal of Physics F: Metal Physics 4 (1974) 1865
work page 1974
- [75]
-
[76]
M. Jahnátek, J. Hafner, M. Krajčí, Shear deformation, ideal strength, and stacking fault formation of fcc metals: A density-functional study of al and cu, Physical Review B—Condensed Matter and Materials Physics 79 (2009) 224103
work page 2009
- [77]
-
[78]
P. Desai, Thermodynamic properties of aluminum, International journal of thermo- physics 8 (1987) 621–638
work page 1987
-
[79]
N. V. Kozyrev, V. V. Gordeev, Thermodynamic properties and equation of state for solid and liquid aluminum, Metals 12 (2022) 1346
work page 2022
-
[80]
B.-Y. Ning, L.-Y. Zhang, An ab initio study of structural phase transitions of crys- talline aluminium under ultrahigh pressures based on ensemble theory, Computational Materials Science 218 (2023) 111960
work page 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.