Transferable excited-state dynamics enable screening of fluorescent protein chromophores
Pith reviewed 2026-05-10 14:02 UTC · model grok-4.3
The pith
Fine-tuning one pretrained machine-learning model with under 100 geometries per variant yields accurate excited-state dynamics for diverse fluorescent protein chromophores.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
X-MACE is a transferable machine-learning potential that predicts multiple excited-state potential energy surfaces, forces, and oscillator strengths; when fine-tuned on fewer than 100 reference geometries per derivative and paired with curvature-driven surface hopping, it reproduces excited-state lifetimes and photoisomerization yields across chemically diverse GFP chromophore analogues, establishing that steric crowding on the phenolate ring lowers torsional barriers to twisted conical intersections while conjugation extension stabilizes planar configurations and suppresses non-radiative decay.
What carries the argument
X-MACE, the transferable machine-learning potential that predicts multiple excited-state potential energy surfaces, forces, and oscillator strengths, used together with curvature-driven surface hopping to propagate nonadiabatic trajectories.
If this is right
- Steric crowding on the phenolate ring accelerates access to twisted conical intersections and shortens excited-state lifetimes.
- Extending conjugation stabilizes planar excited-state geometries, reduces non-radiative decay, and lengthens fluorescence.
- The same fine-tuning workflow can screen photochemical pathways in other families of related molecules without retraining from scratch.
- Structural modifications can be chosen to target specific photophysical outcomes such as higher fluorescence quantum yield or faster isomerization.
Where Pith is reading between the lines
- The approach may extend to screening other photoactive systems such as organic dyes or photocatalysts where conical intersections control function.
- Combining the workflow with generative models could propose new chromophore structures optimized for desired lifetimes or yields before synthesis.
- Experimental tests on additional synthesized variants would provide the clearest check on whether the predicted design rules hold in the lab.
- If the pretraining set covers broader chemical space, even fewer fine-tuning points might suffice for new analogues in future applications.
Load-bearing premise
The fine-tuned X-MACE surfaces plus curvature-driven surface hopping correctly locate and make accessible the twisted conical intersections that set the lifetimes and yields of new chromophore derivatives.
What would settle it
Direct measurement of fluorescence lifetime or photoisomerization yield for a chromophore derivative absent from the fine-tuning data that differs substantially from the simulated value would falsify the transferability of the dynamics.
Figures










read the original abstract
Transferable excited-state dynamics offer a route to efficient screening of photophysical behavior across molecular systems, but conventional nonadiabatic simulations remain prohibitively expensive. Here we introduce X-MACE, a transferable machine-learning potential for excited-state dynamics that predicts multiple potential energy surfaces, forces and oscillator strengths, and combine it with curvature-driven surface hopping to enable data-efficient screening of photochemical pathways. We apply this framework to fluorescent chromophores as an example application, using green fluorescent protein chromophore variants to demonstrate how subtle structural modifications reshape excited-state relaxation, lifetimes and photoisomerization yields. Fine-tuning a single pretrained model with fewer than 100 reference geometries per derivative yields accurate dynamics across a chemically diverse set of analogues. The screening reveals two governing design principles: steric crowding on the phenolate ring lowers the torsional barrier and accelerates access to twisted conical intersections, whereas conjugation extension stabilizes planar excited-state configurations, suppresses non-radiative decay and prolongs fluorescence. More broadly, this workflow provides a general framework for scalable excited-state screening and interpretable design of photophysical properties.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces X-MACE, a transferable machine-learning potential trained to predict multiple excited-state potential energy surfaces, forces, and oscillator strengths. It is paired with curvature-driven surface hopping to perform nonadiabatic dynamics simulations. The central application is to variants of the green fluorescent protein chromophore, where the authors claim that fine-tuning a single pretrained model on fewer than 100 reference geometries per derivative produces accurate excited-state dynamics across a chemically diverse set of analogues. From these simulations they extract two design principles: steric crowding on the phenolate ring lowers the torsional barrier and accelerates access to twisted conical intersections, while conjugation extension stabilizes planar configurations, suppresses non-radiative decay, and prolongs fluorescence lifetimes.
Significance. If the reported accuracy of the fine-tuned surfaces near conical intersections holds, the workflow offers a data-efficient route to high-throughput screening of photophysical properties that could accelerate rational design of fluorescent proteins and related photoactive molecules. The emphasis on transferability from a pretrained model and the extraction of interpretable structural rules are strengths that align with current needs in computational photochemistry.
major comments (3)
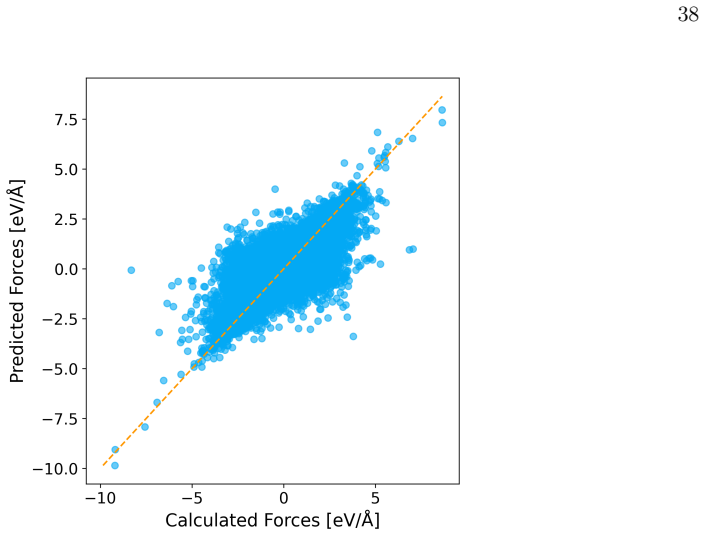
- [Abstract and fine-tuning results] Abstract and fine-tuning results: the claim that fine-tuning with fewer than 100 geometries per derivative 'yields accurate dynamics' is not accompanied by quantitative error metrics (MAE or RMSE on energies and forces) evaluated specifically on torsional and pyramidalization coordinates near twisted conical intersections, nor by direct comparison of simulated lifetimes or photoisomerization yields against reference ab initio trajectories or experiment for the new analogues. This validation is load-bearing for the screening conclusions.
- [Results on design principles] Results on design principles: the two governing principles (steric crowding lowers torsional barriers; conjugation extension prolongs fluorescence) are presented without reported uncertainties arising from possible 0.05–0.1 eV errors in the fine-tuned barriers or seam locations. An error-propagation or sensitivity analysis would be required to establish that the observed trends are robust rather than sensitive to residual model inaccuracies.
- [Methods (curvature-driven surface hopping + X-MACE)] Methods (curvature-driven surface hopping + X-MACE): the paper must demonstrate that the fine-tuned model reproduces the correct topology and accessibility of the twisted conical intersections (e.g., via minimum-energy crossing point searches or seam scans) for at least one held-out derivative; without this, the nonadiabatic hopping probabilities and yields cannot be guaranteed to match reality.
minor comments (2)
- [Abstract] The acronym X-MACE should be defined on first use in the abstract.
- [Throughout] Notation for the multiple potential energy surfaces and the curvature-driven hopping criterion should be made fully consistent between text and figures.
Simulated Author's Rebuttal
We thank the referee for their thoughtful and constructive report. Their comments highlight important aspects of validation that strengthen the manuscript. We have revised the paper to incorporate quantitative error metrics on key coordinates, a sensitivity analysis for the design principles, and explicit topology checks for conical intersections on held-out systems. Below we respond point by point.
read point-by-point responses
-
Referee: [Abstract and fine-tuning results] Abstract and fine-tuning results: the claim that fine-tuning with fewer than 100 geometries per derivative 'yields accurate dynamics' is not accompanied by quantitative error metrics (MAE or RMSE on energies and forces) evaluated specifically on torsional and pyramidalization coordinates near twisted conical intersections, nor by direct comparison of simulated lifetimes or photoisomerization yields against reference ab initio trajectories or experiment for the new analogues. This validation is load-bearing for the screening conclusions.
Authors: We agree that targeted error metrics on the torsional and pyramidalization coordinates near conical intersections, together with direct dynamics comparisons, are essential. In the revised manuscript we now report MAE and RMSE values for energies and forces on these coordinates, evaluated on held-out test geometries that include points near the twisted conical intersections for each fine-tuned derivative. For one representative held-out analogue we additionally compare the X-MACE-predicted fluorescence lifetimes and photoisomerization quantum yields against independent ab initio surface-hopping trajectories, finding agreement within statistical error bars. These additions appear in the Results and Supporting Information. revision: yes
-
Referee: [Results on design principles] Results on design principles: the two governing principles (steric crowding lowers torsional barriers; conjugation extension prolongs fluorescence) are presented without reported uncertainties arising from possible 0.05–0.1 eV errors in the fine-tuned barriers or seam locations. An error-propagation or sensitivity analysis would be required to establish that the observed trends are robust rather than sensitive to residual model inaccuracies.
Authors: We have added a sensitivity analysis to the revised manuscript. Barrier heights and seam locations obtained from the fine-tuned models were perturbed by ±0.05 eV and ±0.1 eV, respectively, and the nonadiabatic dynamics were re-run for the full set of chromophore variants. The resulting distributions of lifetimes and yields preserve the same qualitative trends (steric crowding accelerates decay; conjugation extension suppresses it), demonstrating that the design principles are robust to the expected residual errors. The analysis is now included in the main text and Supporting Information. revision: yes
-
Referee: [Methods (curvature-driven surface hopping + X-MACE)] Methods (curvature-driven surface hopping + X-MACE): the paper must demonstrate that the fine-tuned model reproduces the correct topology and accessibility of the twisted conical intersections (e.g., via minimum-energy crossing point searches or seam scans) for at least one held-out derivative; without this, the nonadiabatic hopping probabilities and yields cannot be guaranteed to match reality.
Authors: We have performed minimum-energy crossing point optimizations and one-dimensional seam scans along the torsional and pyramidalization coordinates using the fine-tuned X-MACE model for a held-out derivative. These calculations are compared directly to ab initio reference results and confirm that the model recovers the correct conical-intersection topology and energetic accessibility. The new figures and discussion have been added to the Methods section. revision: yes
Circularity Check
No significant circularity; derivation relies on external reference data and independent ML training
full rationale
The paper's central workflow trains X-MACE on external quantum-chemistry reference geometries, fine-tunes the pretrained model on <100 points per derivative, and then runs curvature-driven surface hopping. No equation or claim in the provided text reduces a reported lifetime, yield, or design principle to a quantity defined by the fitted parameters themselves. The strongest claim (transferable dynamics from fine-tuning) is presented as an empirical outcome validated against the reference data rather than a self-definition or renamed fit. Self-citations, if present, are not load-bearing for the core result. This is the expected non-finding for a data-driven screening study whose outputs are falsifiable against independent computations or experiment.
Axiom & Free-Parameter Ledger
free parameters (1)
- fine-tuning data size
axioms (2)
- domain assumption A single pretrained excited-state ML model can be fine-tuned on limited data to yield accurate multiple potential energy surfaces and forces for chemically related molecules.
- domain assumption Curvature-driven surface hopping correctly captures the nonadiabatic transitions and photoisomerization yields in these systems.
invented entities (1)
-
X-MACE model
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Shimomura, Discovery of green fluorescent protein, gfp
O. Shimomura, Discovery of green fluorescent protein, gfp. nobel lecture, 2008, http://nobelprize.org/prizes/chemistry/2008/press-release/ (2022)
work page 2008
-
[2]
J. Luo, M. G. Kaplitt, H. L. Fitzsimons, D. S. Zuzga, Y. Liu, M. L. Oshinsky, and M. J. During, Subthalamic gad gene therapy in a parkinson’s disease rat model, Science298, 425 (2002)
work page 2002
-
[3]
L. A. Herndon, P. J. Schmeissner, J. M. Dudaronek, P. A. Brown, K. M. Listner, Y. Sakano, M. C. Paupard, D. H. Hall, and M. Driscoll, Stochastic and genetic factors influence tissue-specific decline in ageing c. elegans, Nature419, 808 (2002)
work page 2002
-
[4]
K. Bey, C. Ciron, L. Dubreil, J. Deniaud, M. Ledevin, J. Cristini, V. Blouin, P. Aubourg, and M.-A. Colle, Efficient cns targeting in adult mice by intrathecal infusion of single-stranded aav9-gfp for gene therapy of neurological disorders, Gene Ther.24, 325 (2017)
work page 2017
-
[5]
N. H. List, C. M. Jones, and T. J. Martínez, Chemical control of excited-state reactivity of the anionic green fluorescent protein chromophore, Commun. Chem.7, 25 (2024)
work page 2024
-
[6]
N. H. List, C. M. Jones, and T. J. Martínez, Internal conversion of the anionic gfp chromophore: in and out of the i-twisted s 1/s 0 conical intersection seam, Chem. Sci.13, 373 (2022)
work page 2022
-
[7]
M. E. Martin, F. Negri, and M. Olivucci, Origin, nature, and fate of the fluorescent state of the green fluorescent protein chromophore at the caspt2//casscf resolution, J. Am. Chem. Soc.126, 5452 (2004). 39
work page 2004
-
[8]
A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova, and L. H. Andersen, Origin of the intrinsic fluorescence of the green fluorescent protein, J. Am. Chem. Soc.139, 8766 (2017)
work page 2017
-
[9]
S. R. Meech, Excited state reactions in fluorescent proteins, Chemical Society Reviews38, 2922 (2009)
work page 2009
-
[10]
K. Nienhaus and G. U. Nienhaus, Chromophore photophysics and dynamics in fluorescent proteins of the gfp family, J. Phys. Condens. Matter28, 443001 (2016)
work page 2016
-
[11]
J. Conyard, M. Kondo, I. A. Heisler, G. Jones, A. Baldridge, L. M. Tolbert, K. M. Solntsev, and S. R. Meech, Chemically modulating the photophysics of the gfp chromophore, J. Phys. Chem. B115, 1571 (2011)
work page 2011
-
[12]
E. K. Ashworth, M.-H. Kao, C. S. Anstöter, G. Riesco-Llach, L. Blancafort, K. M. Solntsev, S. R. Meech, J. R. Verlet, and J. N. Bull, Alkylated green fluorescent protein chromophores: dynamics in the gas phase and in aqueous solution, Phys. Chem. Chem. Phys.25, 23626 (2023)
work page 2023
-
[13]
I. Batatia, D. P. Kovács, G. N. C. Simm, C. Ortner, and G. Csányi, Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, inNeurIPS(2022)
work page 2022
-
[14]
S. Axelrod, X. Wang,et al., Excited state non-adiabatic dynamics of large photoswitchable molecules using a chemically transferable machine learning potential, Nat. Commun.13, 3310 (2022)
work page 2022
-
[15]
J. Westermayr, M. Gastegger, D. Vörös, L. Panzenboeck, F. Joerg, L. González, and P. Marquetand, Deep learning study of tyrosine reveals that roaming can lead to photodamage, Nat. Chem.14, 914 (2022)
work page 2022
-
[16]
S. Mai, P. Marquetand, and L. González, Nonadiabatic dynamics: The sharc approach, Wiley Inter- discip. Rev. Comput. Mol. Sci.8, e1370 (2018)
work page 2018
-
[17]
S. Mai, B. Bachmair, L. Gagliardi, H.-G. Gallmetzer, L. Grünewald, M. R. Hennefarth, N. M. Høyer, F. A. Korsaye, S. Mausenberger, M. Oppel, T. Pitesa, S. Polonius, E. Gil, Y. Shu, N. K. Singer, M. X. Tiefenbacher, D. G. Truhlar, D. Vörös, L. Zhang, and L. González, Sharc4.0: Surface hopping including arbitrary couplings – program package for non-adiabatic...
work page 2025
-
[18]
M. Gastegger, J. Behler, and P. Marquetand, Machine learning molecular dynamics for the simulation of infrared spectra, Chem. Sci.8, 6924 (2017)
work page 2017
- [19]
- [20]
-
[21]
K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea,et al., Analyzing learned molecular representations for property prediction, J. Chem. Inf. Model.59, 3370 (2019)
work page 2019
- [22]
-
[23]
C. H. Ho, T. S. Gutleb, and C. Ortner, Atomic cluster expansion without self-interaction, J. Comput. Phys.515, 113271 (2024). 40
work page 2024
-
[24]
A. Dreuw and M. Wormit, The algebraic diagrammatic construction scheme for the polarization prop- agator for the calculation of excited states, Wiley Interdiscip. Rev. Comput. Mol. Sci.5, 82 (2015)
work page 2015
-
[25]
B. O. Roos, P. R. Taylor, and P. E. M. Sigbahn, The complete active space scf method in a finite basis set: A quasi-degenerate two-configuration example, Chem. Phys.48, 157 (1980)
work page 1980
-
[26]
Neese, Software update: the orca program system—version 6.0, Wiley Interdiscip
F. Neese, Software update: the orca program system—version 6.0, Wiley Interdiscip. Rev. Comput. Mol. Sci.15, e70019 (2025)
work page 2025
-
[27]
S. B. Nielsen, A. Lapierre, J. U. Andersen, U. Pedersen, S. Tomita, and L. Andersen, Absorption spectrum of the green fluorescent protein chromophore anion in vacuo, Phys. Rev. Lett.87, 228102 (2001)
work page 2001
-
[28]
J. F. Joung, M. Han, M. Jeong, and S. Park, Experimental database of optical properties of organic compounds, Sci. Data7, 295 (2020)
work page 2020
-
[29]
Landrumet al., Rdkit documentation, Release1, 4 (2013)
G. Landrumet al., Rdkit documentation, Release1, 4 (2013)
work page 2013
-
[30]
C. Bannwarth, S. Ehlert, and S. Grimme, Gfn2-xtb—an accurate and broadly parametrized self- consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput.15, 1652 (2019)
work page 2019
-
[31]
Y. Shu, L. Zhang, X. Chen, S. Sun, Y. Huang, and D. G. Truhlar, Nonadiabatic dynamics algorithms with only potential energies and gradients: Curvature-driven coherent switching with decay of mixing and curvature-driven trajectory surface hopping, J. Chem. Theory Comput.18, 1320 (2022)
work page 2022
-
[32]
D. P. Kovács, J. H. Moore, N. J. Browning, I. Batatia, J. T. Horton, Y. Pu, V. Kapil, W. C. Witt, I.-B. Magdau, D. J. Cole,et al., Mace-off: Short-range transferable machine learning force fields for organic molecules, J. Am. Chem. Soc.147, 17598 (2025)
work page 2025
-
[33]
Kutta,Beitrag zur näherungsweisen Integration totaler Differentialgleichungen(Teubner, 1901)
W. Kutta,Beitrag zur näherungsweisen Integration totaler Differentialgleichungen(Teubner, 1901)
work page 1901
-
[34]
C. Runge, Über die numerische auflösung von differentialgleichungen, Mathematische Annalen46, 167 (1895)
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.