A Theoretical Investigation of the Thermal and Photochemical Mechanisms of Ethylbenzene Dehydrogenation on Rutile TiO₂(110)
Pith reviewed 2026-05-07 11:47 UTC · model grok-4.3
The pith
Ethylbenzene converts to styrene on TiO2 via proton-coupled electron transfer on clean surfaces but switches to direct hydrogen transfer on oxidized ones, with 257 nm light bypassing high ground-state barriers.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
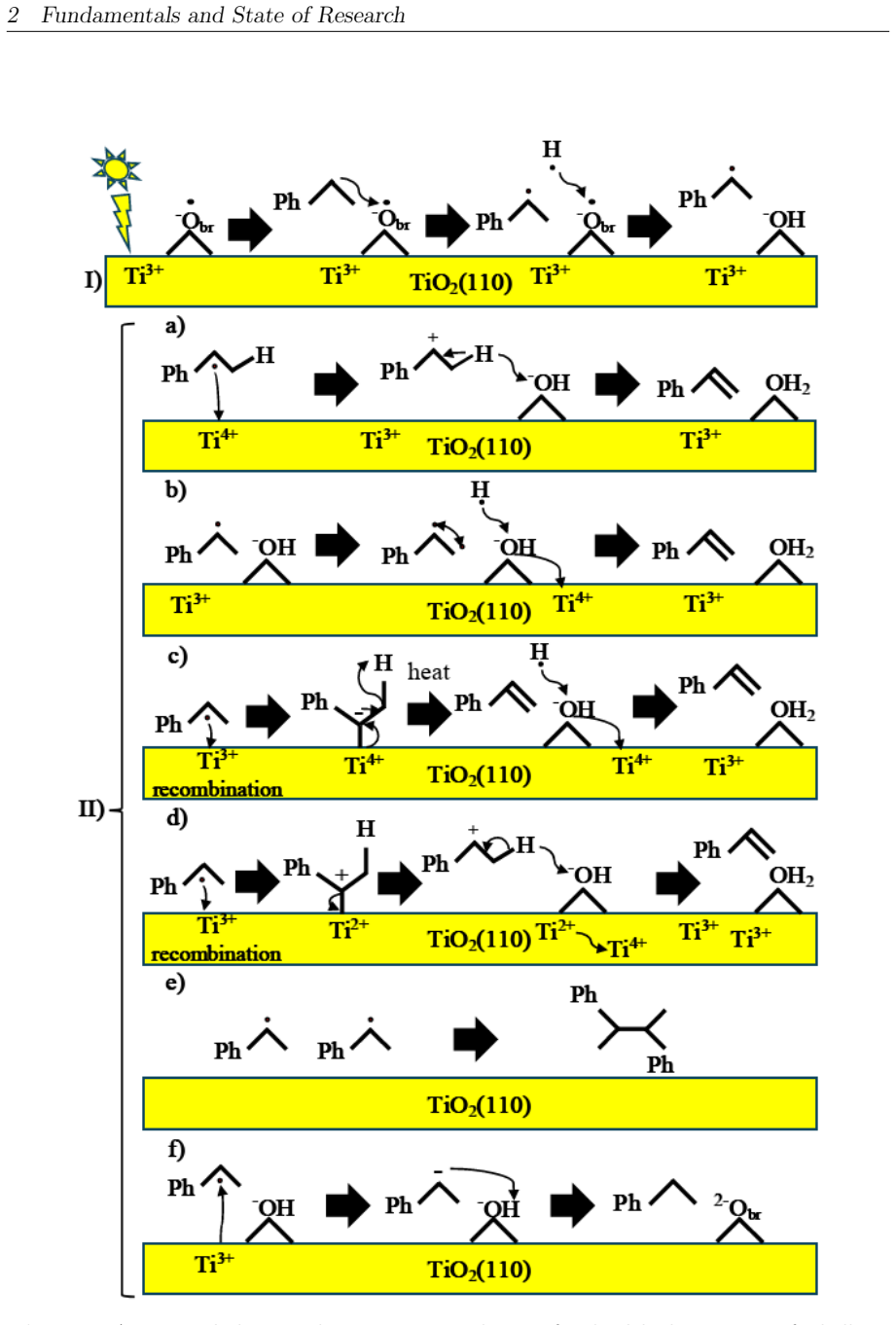
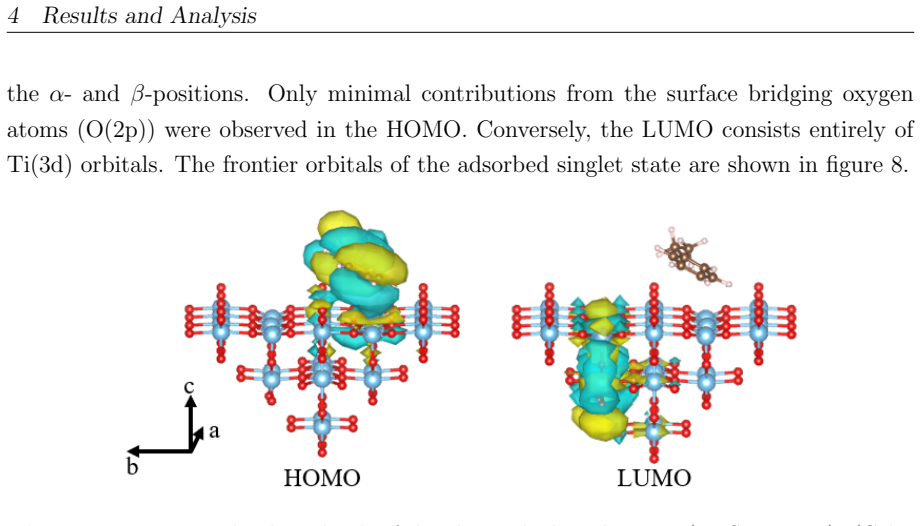
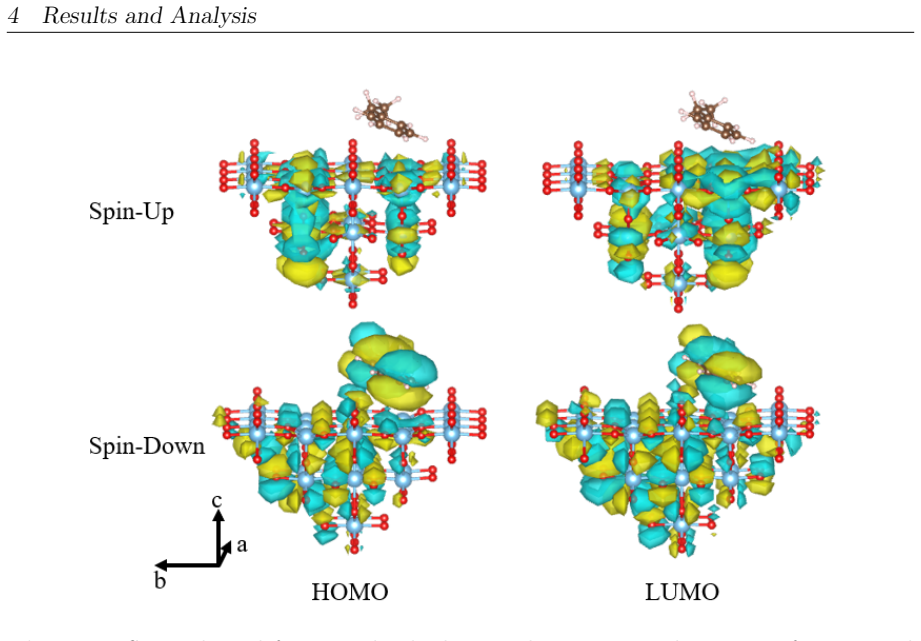
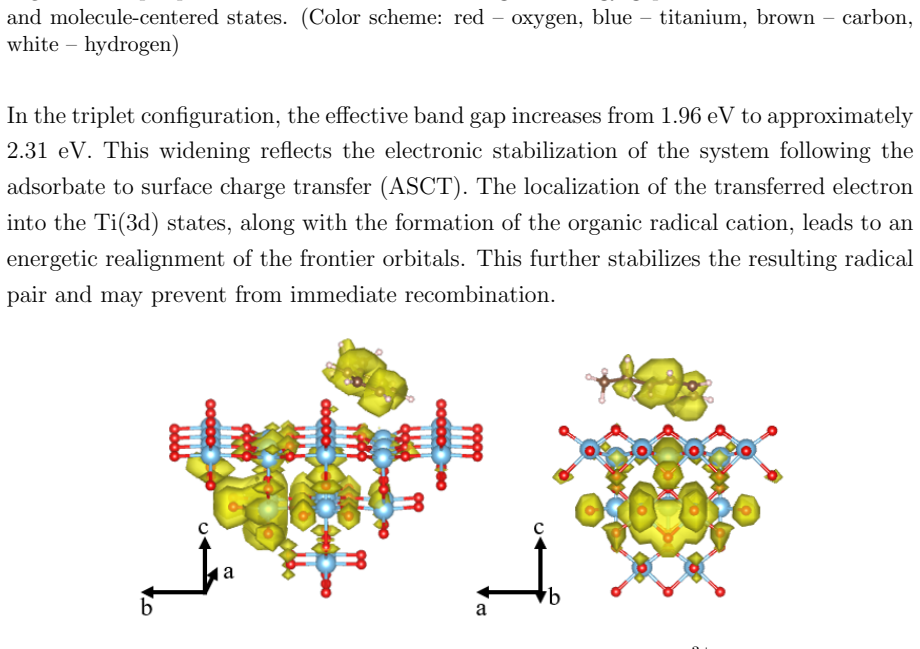
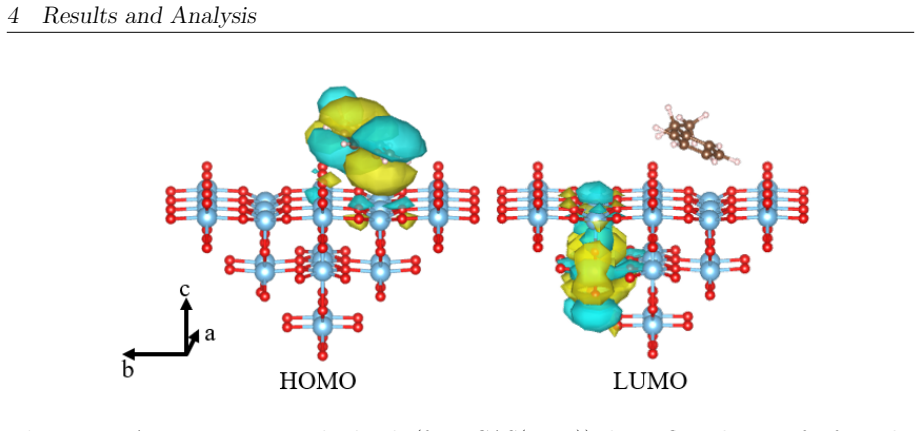
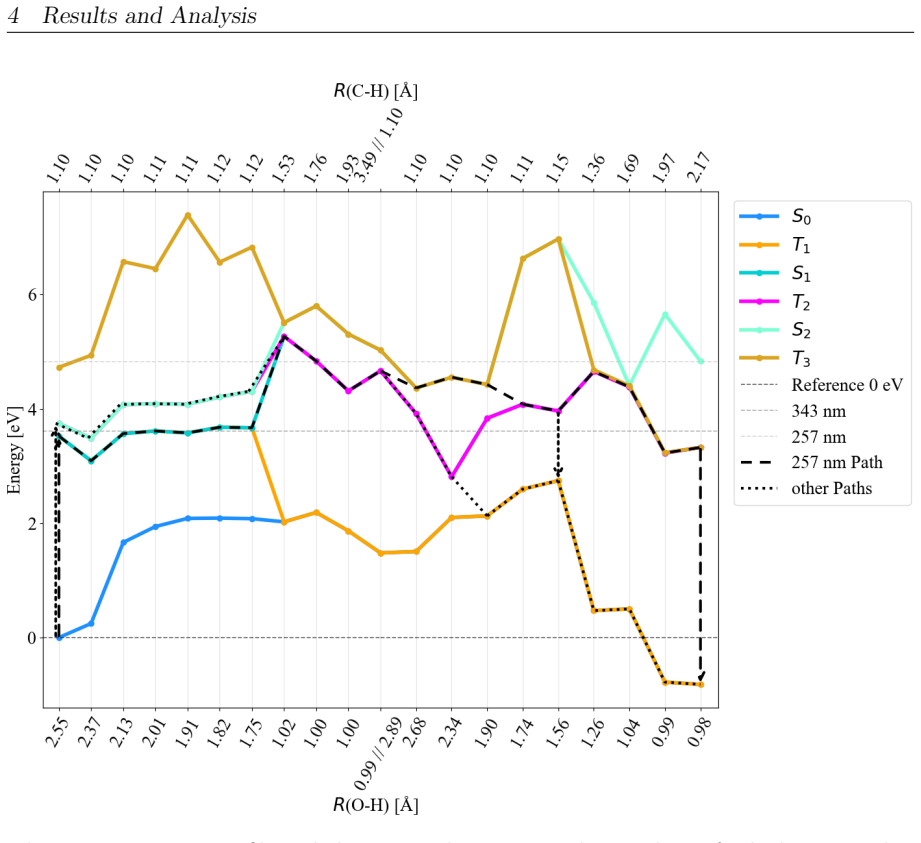
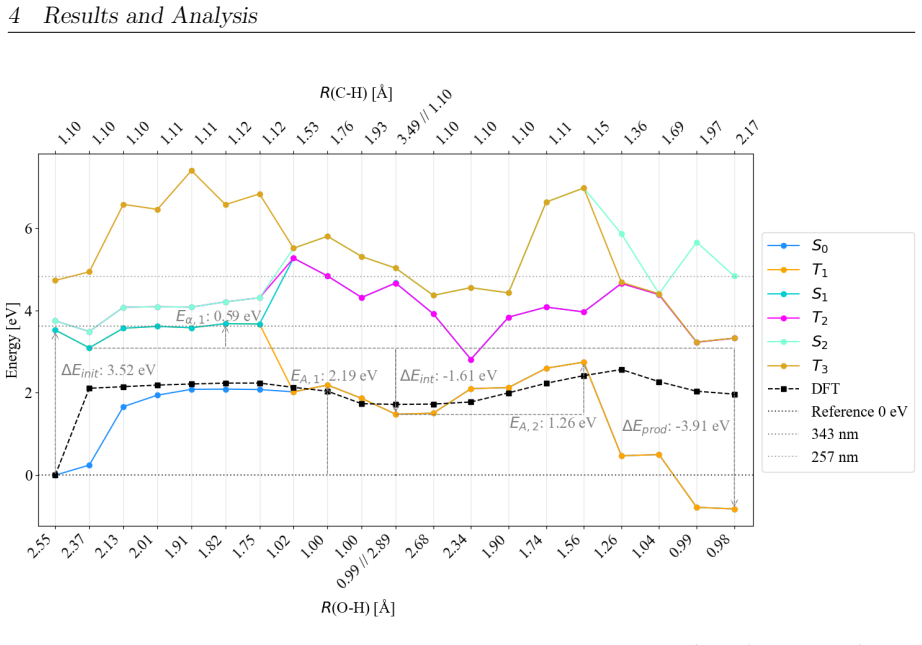
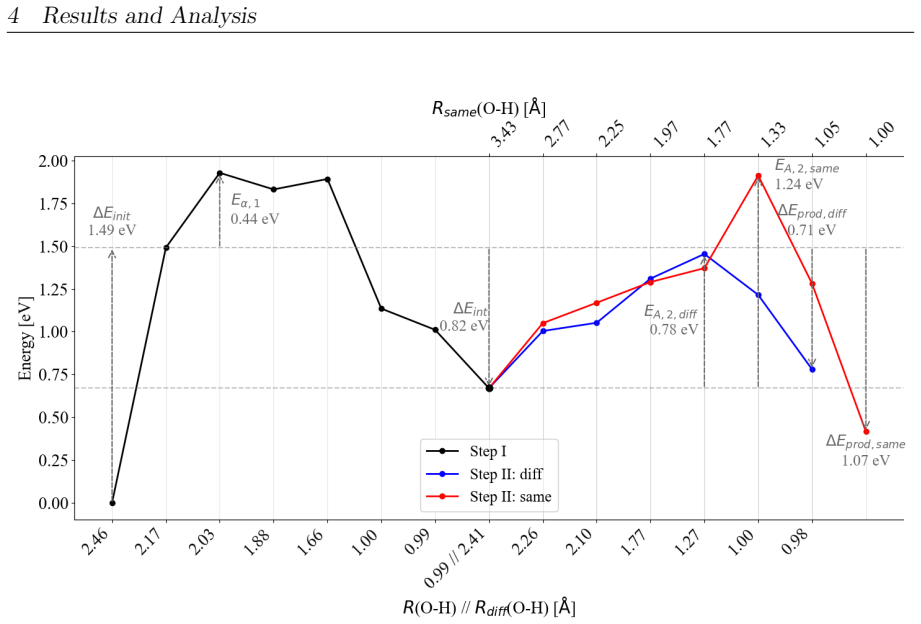
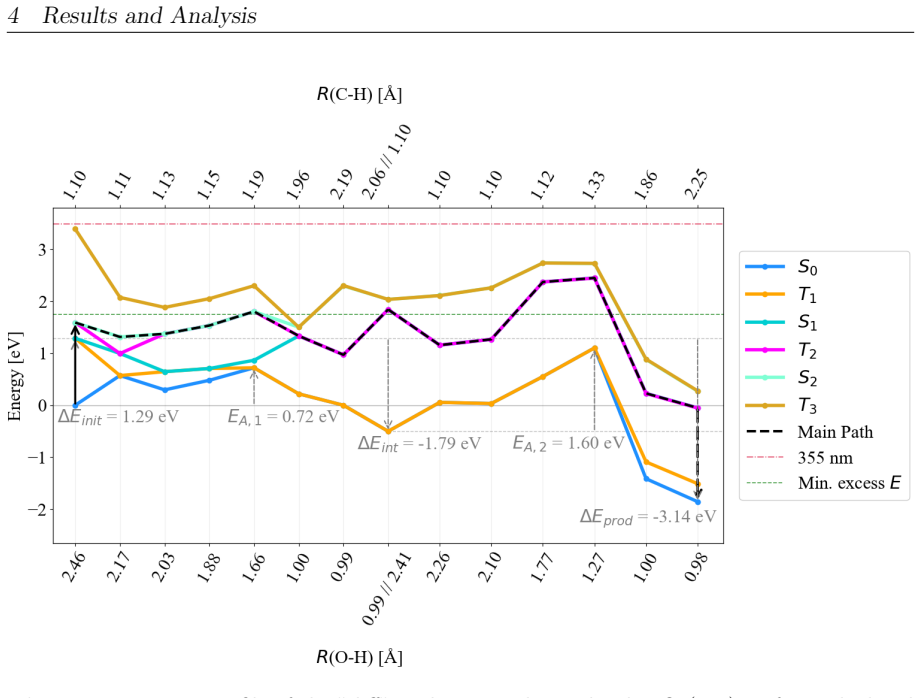
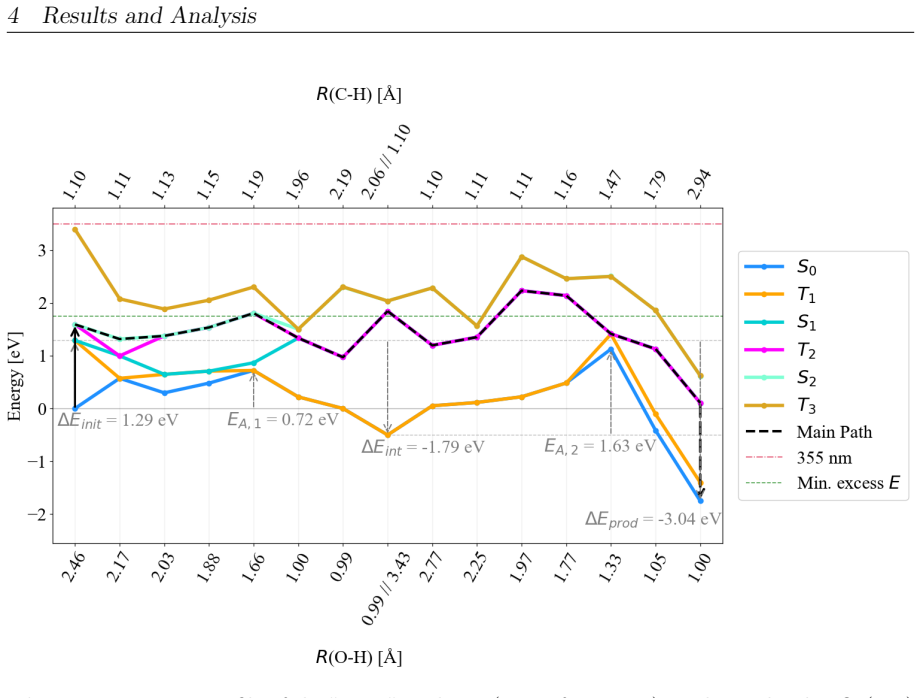
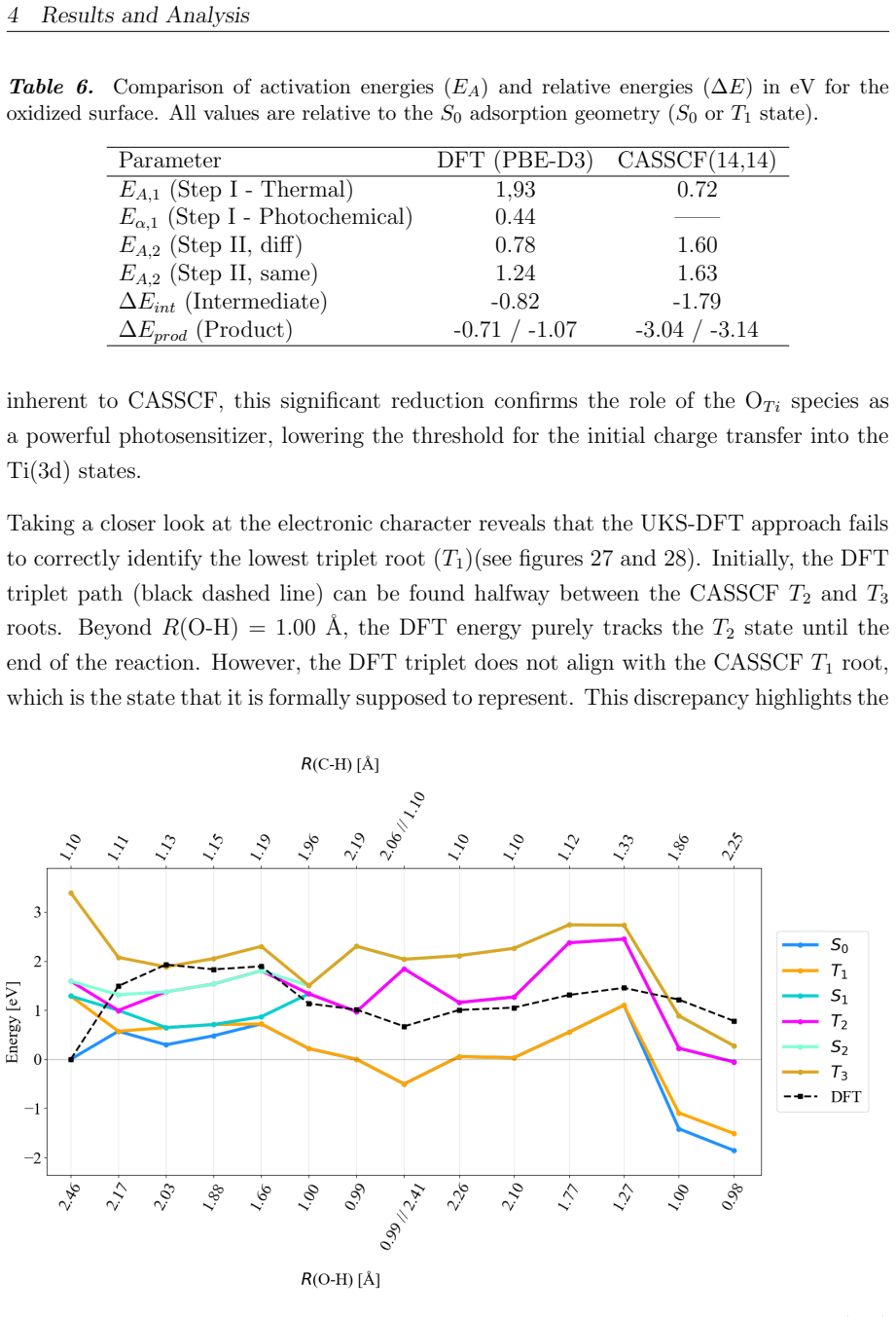
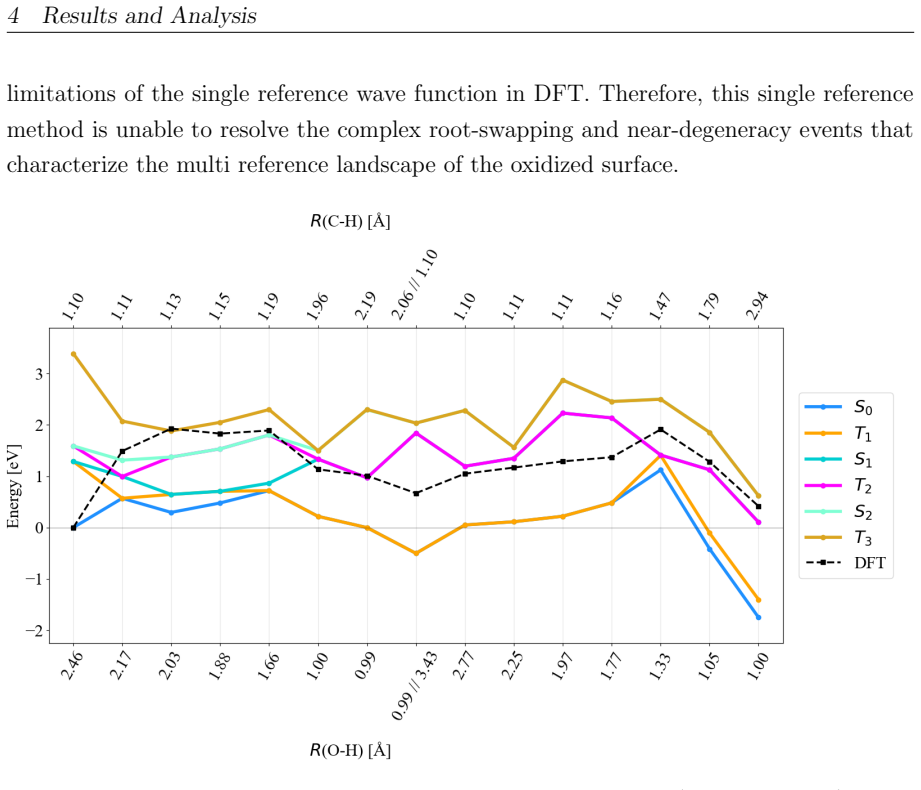
On the stoichiometric surface both thermal and photochemical pathways are dominated by proton-coupled electron transfer. 343 nm irradiation leads to rapid relaxation into the ground state where high kinetic barriers persist, whereas 257 nm excitation enables the system to persist in higher excited states allowing the reaction to bypass the rate-determining ground-state barrier. On oxidized surfaces pre-adsorbed oxygen radicals enable direct hydrogen atom transfer which is more efficient than proton-coupled electron transfer on reduced surfaces.
What carries the argument
Proton-coupled electron transfer on stoichiometric surfaces versus direct hydrogen atom transfer enabled by pre-adsorbed oxygen radicals on oxidized surfaces, with persistence in higher excited electronic states (S1/T2) under 257 nm light determining whether the ground-state barrier can be avoided.
If this is right
- 257 nm light improves efficiency by allowing the reaction to avoid the ground-state rate-determining step through excited-state persistence.
- Oxidized surfaces increase styrene yield through the more efficient direct hydrogen atom transfer pathway.
- Multi-reference quantum methods are required to properly describe the radical intermediates and excited states in these surface reactions.
- Photocatalysis on TiO2 offers a route to styrene production under milder conditions than high-temperature industrial processes.
Where Pith is reading between the lines
- Similar shifts from coupled proton-electron to direct hydrogen transfer with surface oxidation could appear in other dehydrogenation reactions on metal oxides.
- Tuning light wavelength to match specific excited states might control yields or selectivity in related photocatalytic conversions on oxide surfaces.
- Testing the mechanisms on other crystal faces or with substituted reactants would reveal how general the oxygen scavenger effect is.
Load-bearing premise
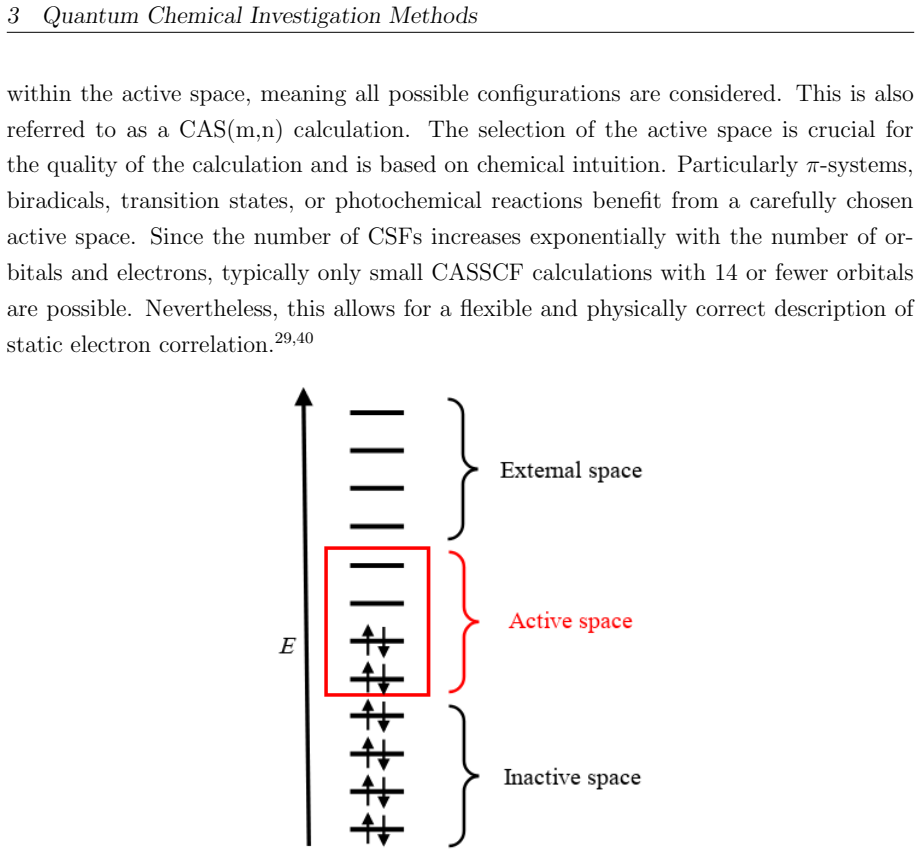
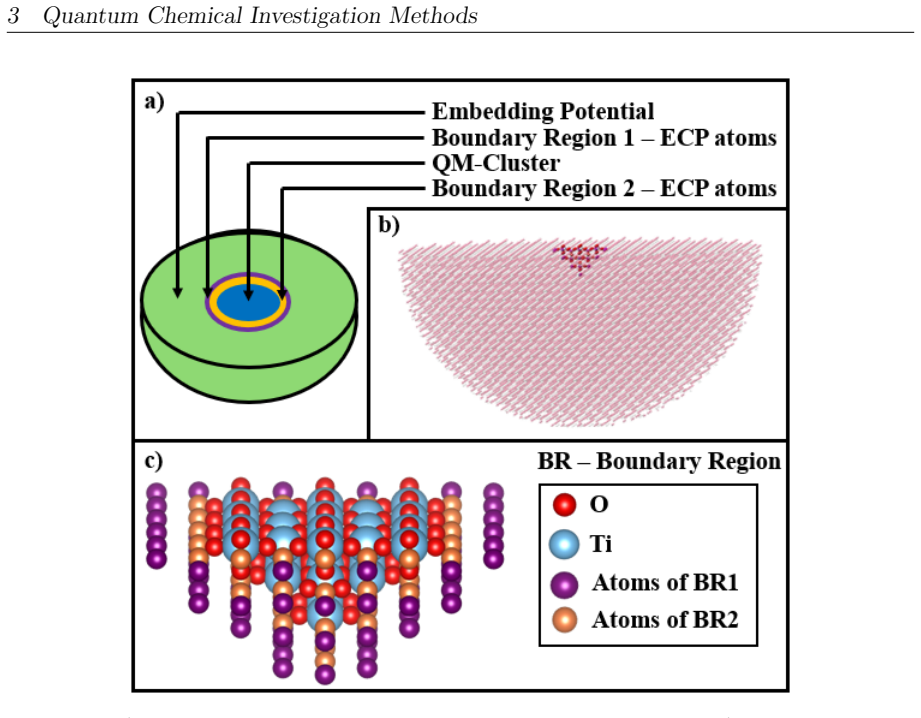
The DFT-PBE-D3 and SA-CASSCF calculations with the chosen surface models accurately capture the actual energy barriers, electronic states, and radical intermediates without major errors from the approximations or incomplete active spaces.
What would settle it
Experimental measurements showing no increase in styrene yield under 257 nm light compared to 343 nm or no yield improvement when oxygen is pre-adsorbed on the surface would contradict the assigned mechanisms and excited-state role.
Figures



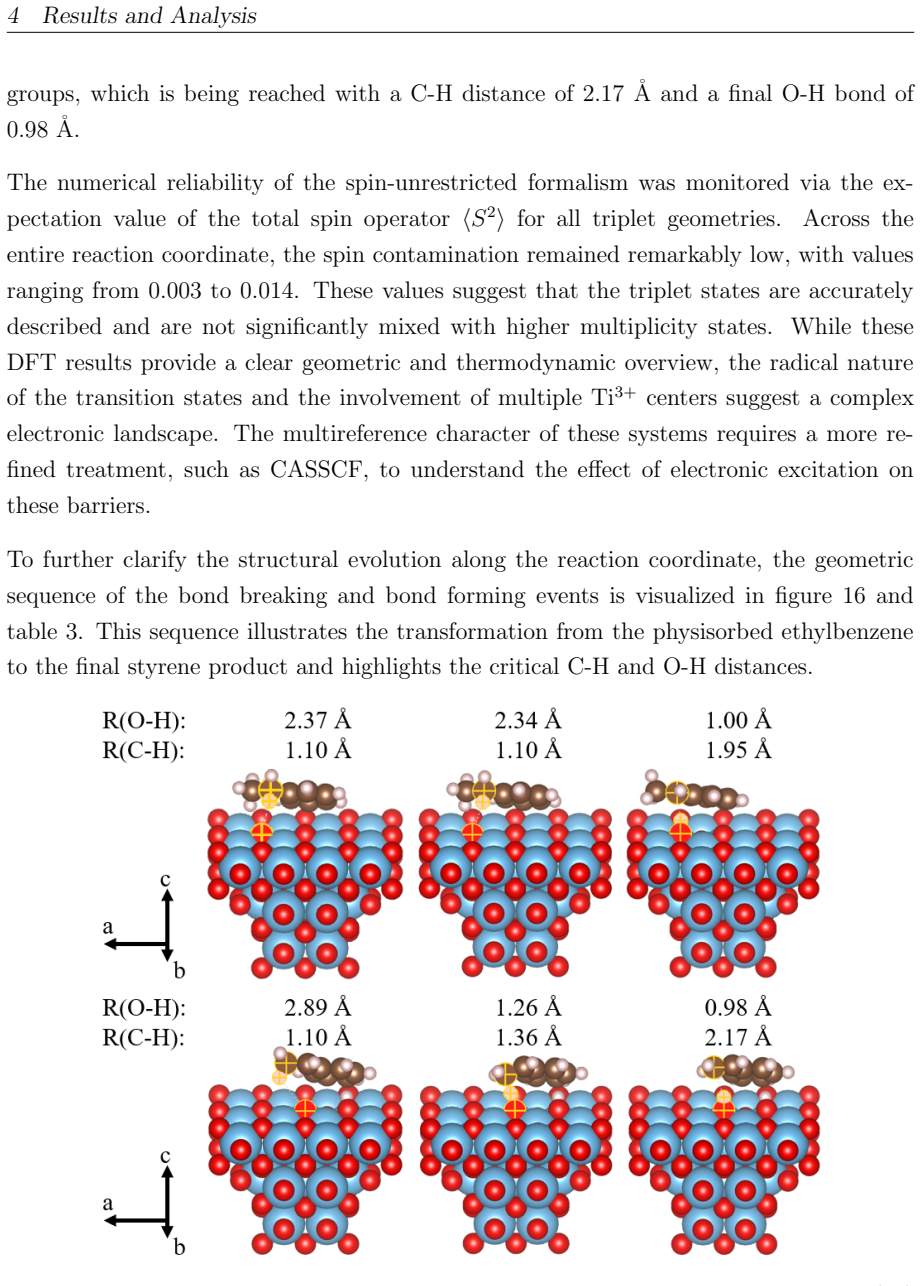
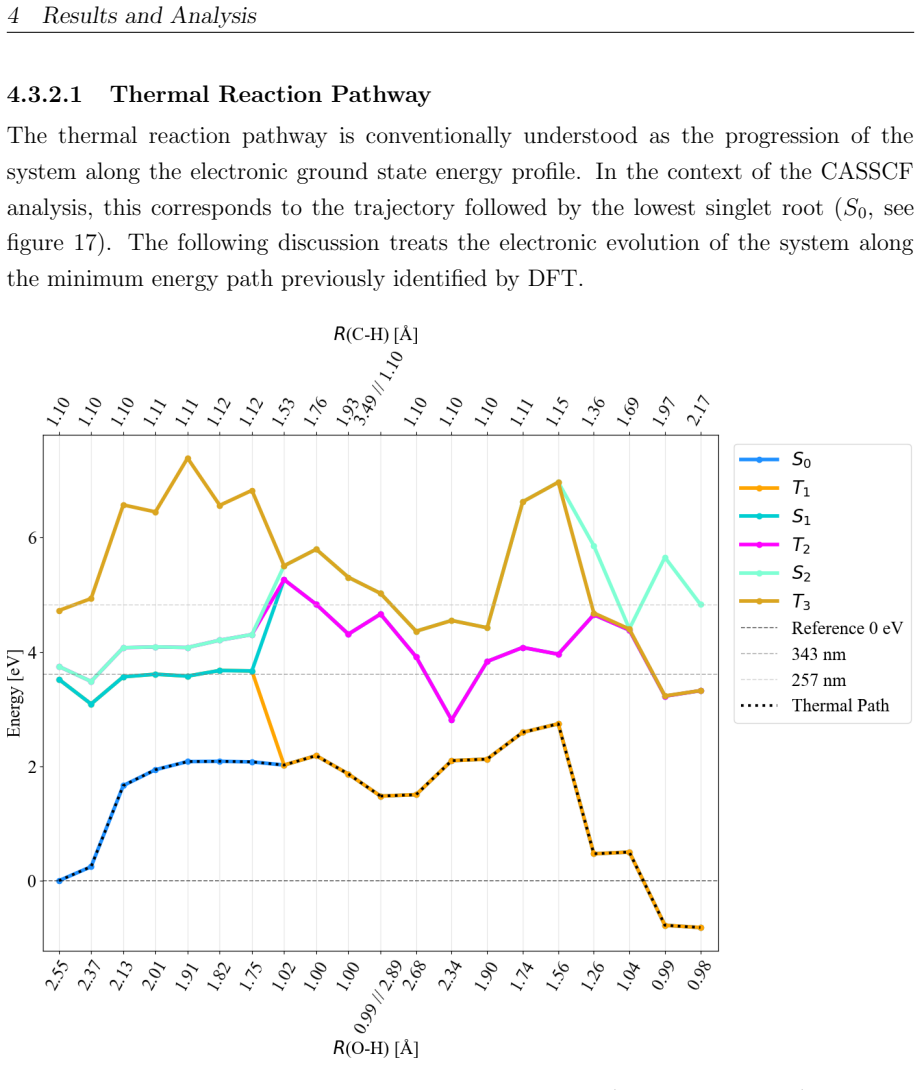
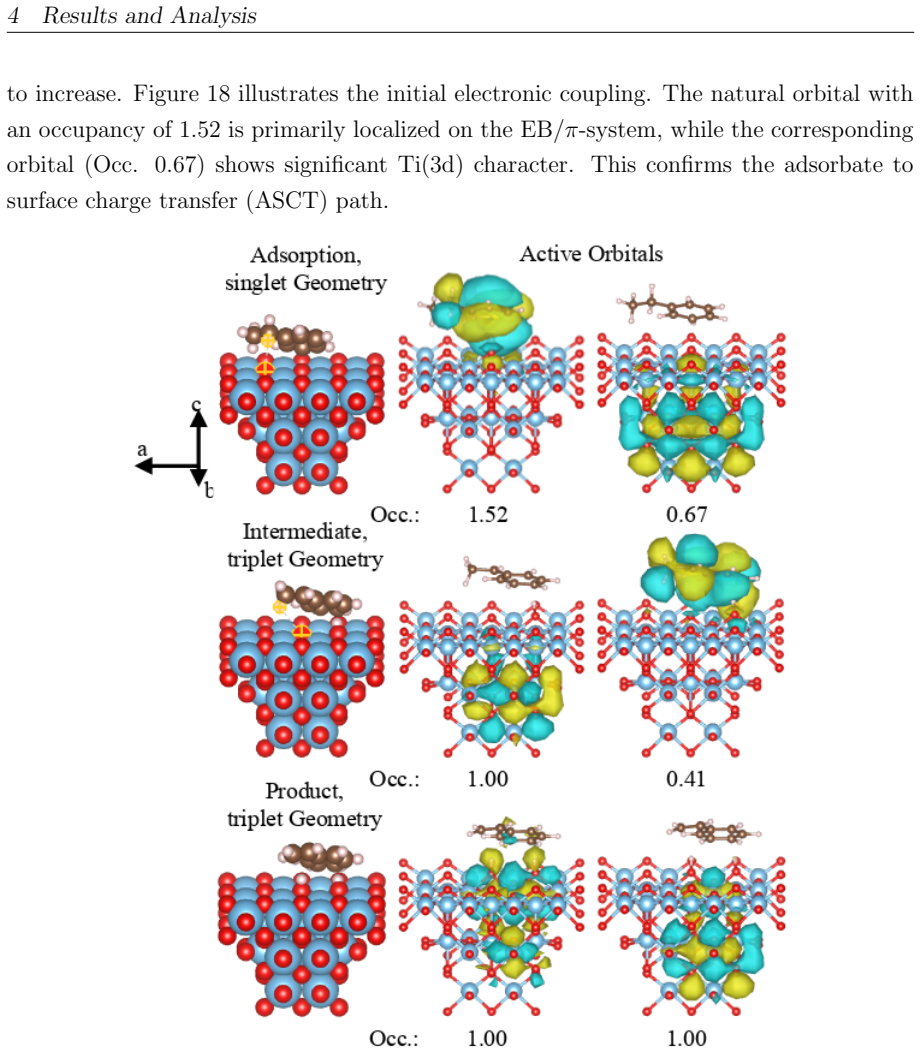
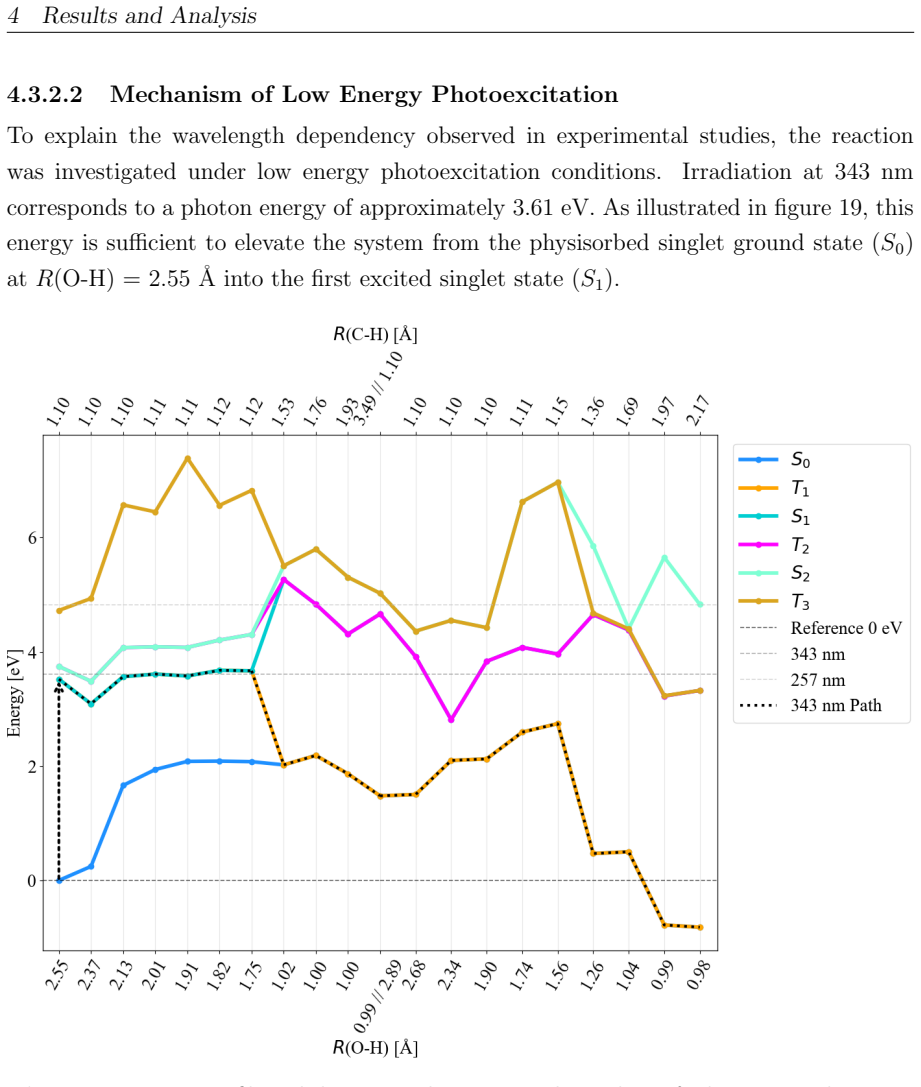

read the original abstract
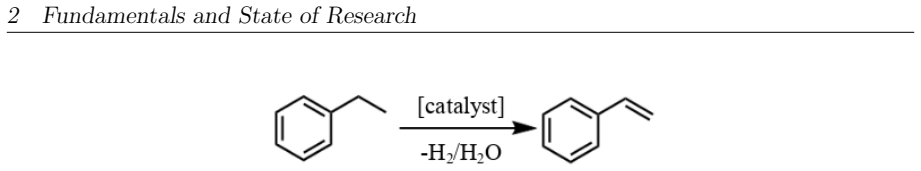
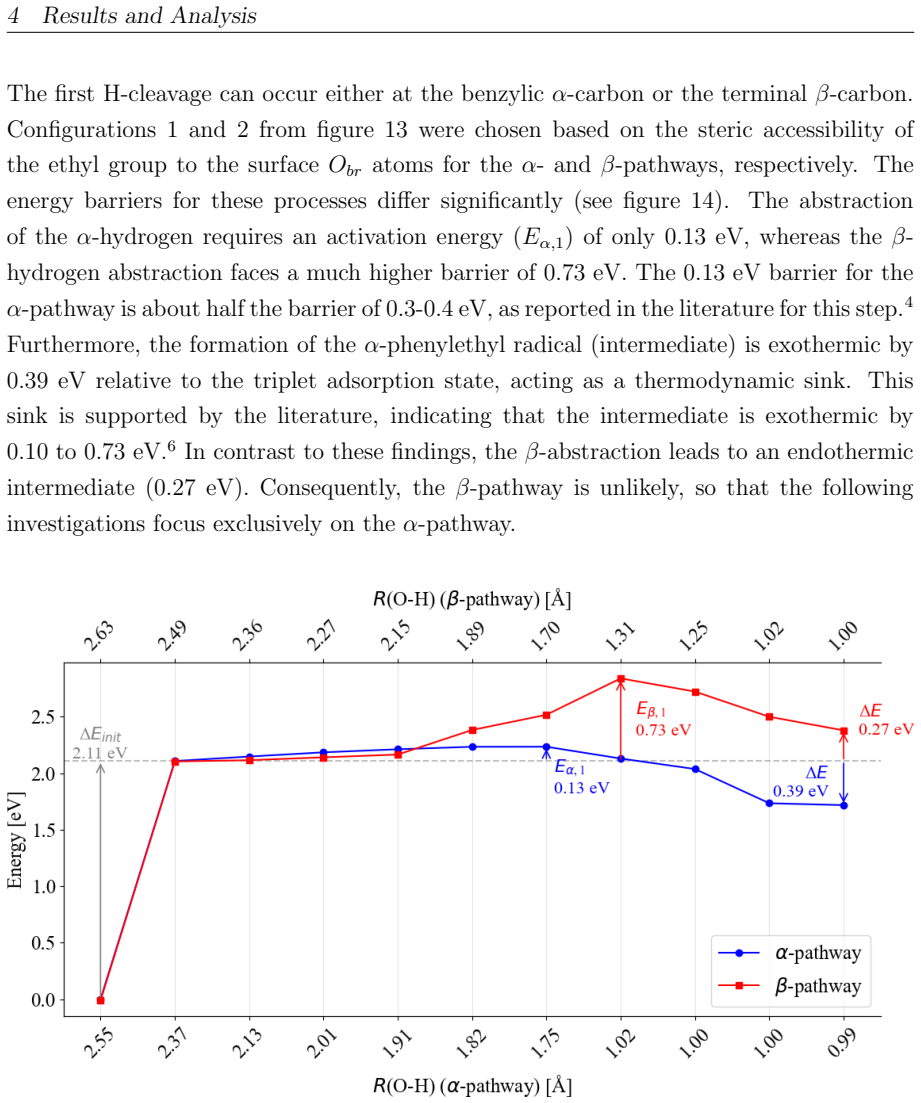
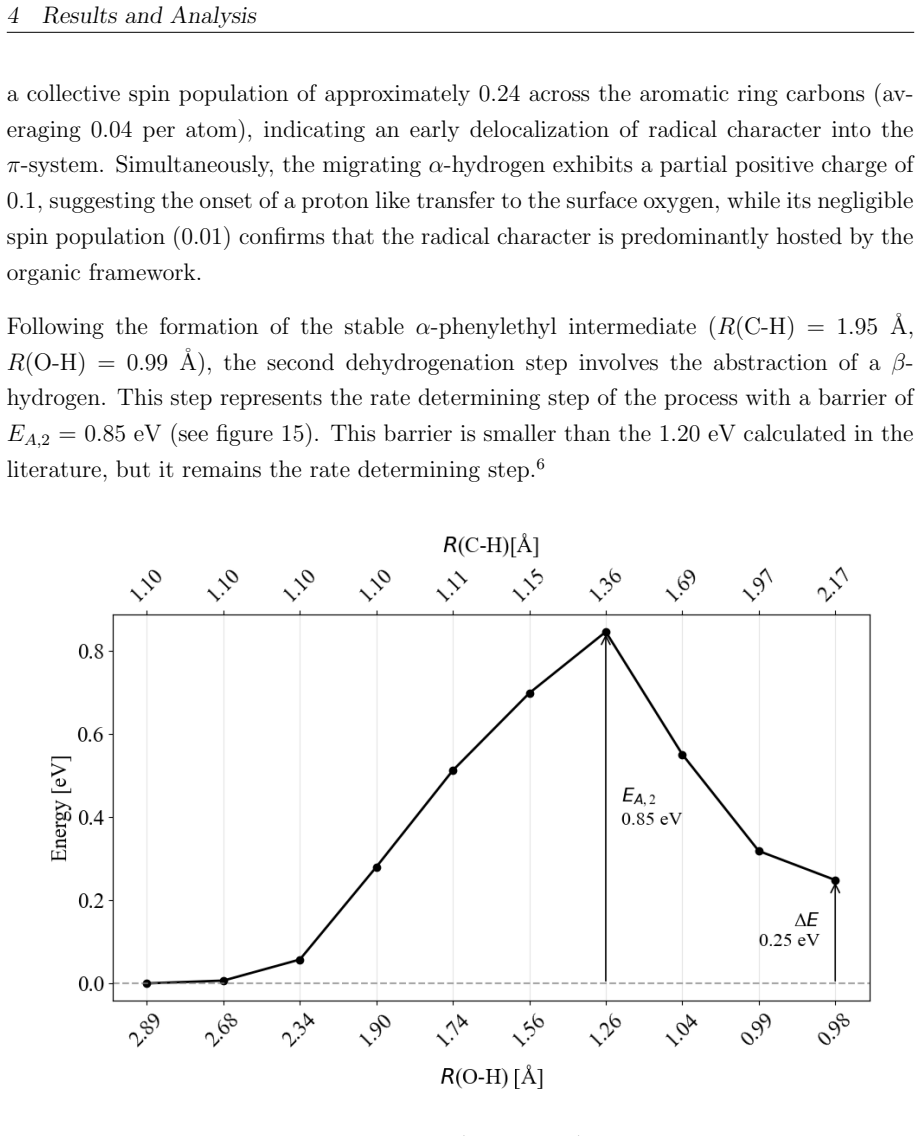
This master's thesis investigates the thermal and photochemical dehydrogenation of ethylbenzene (EB) to styrene on the rutile TiO$_{2}$(110) surface. A dual-methodological quantum chemical approach is used for this investigation. While industrial styrene production is energy-intensive, photocatalysis on semiconductor materials offers a promising alternative under significantly milder conditions. To elucidate the underlying mechanisms, this study employs density functional theory (DFT-PBE-D3) for geometry optimization and high-level multi-reference methods (SA-CASSCF) to accurately describe the electronic complexity of excited states and radical intermediates. The investigation reveals that, on the stoichiometric surface, both thermal and photochemical pathways are dominated by proton-coupled electron transfer (PCET). The wavelength dependence observed in the literature is explained by how the system navigates electronic manifolds. 343 nm irradiation leads to rapid relaxation into the ground state, where high kinetic barriers persist. In contrast, 257 nm excitation enables the system to persist in higher excited states (S1/T2). This allows the reaction to bypass the rate-determining ground-state barrier. Furthermore, the study demonstrates that surface oxidation causes a fundamental mechanistic shift. On oxidized surfaces, pre-adsorbed oxygen radicals (O$_{Ti}$) enable direct hydrogen atom transfer (HAT), which is more efficient than PCET on reduced surfaces. This "hydrogen scavenger" effect explains the significant increase in styrene yield. This work underscores the necessity of multi-reference treatments for complex surface reactions and provides a fundamental understanding of how surface stoichiometry and photon energy govern photocatalytic efficiency.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript is a theoretical study of ethylbenzene dehydrogenation to styrene on rutile TiO2(110). Using DFT-PBE-D3 for ground-state structures and SA-CASSCF for excited states and radicals, it concludes that proton-coupled electron transfer (PCET) dominates both thermal and photochemical pathways on the stoichiometric surface. Wavelength dependence is attributed to persistence in S1/T2 states at 257 nm (bypassing the ground-state barrier) versus rapid relaxation at 343 nm. On oxidized surfaces, pre-adsorbed O_Ti radicals enable direct hydrogen atom transfer (HAT), which is more efficient than PCET and accounts for higher experimental yields.
Significance. If validated, the work supplies a mechanistic rationale for observed wavelength and surface-oxidation effects in TiO2 photocatalysis, highlighting the necessity of multi-reference methods for radical intermediates and excited-state manifolds. This could guide optimization of milder photocatalytic routes to styrene. The dual-method approach is a positive step toward treating electronic complexity on oxide surfaces, though its impact is limited by the absence of parameter reporting and direct experimental benchmarking.
major comments (3)
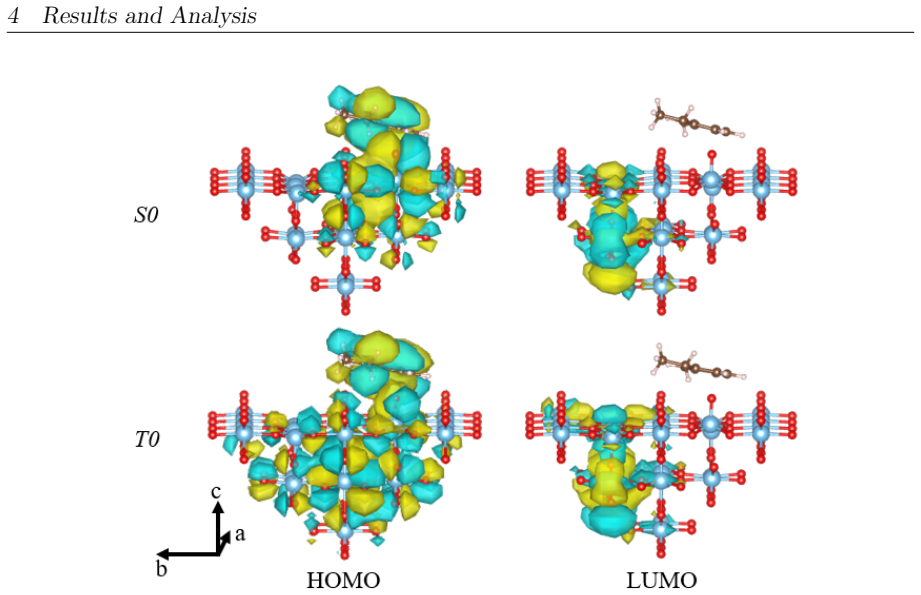
- [Computational Methods] Computational Methods (or equivalent section): The SA-CASSCF calculations are described without specifying the active-space size, orbital selection (e.g., inclusion of Ti 3d, O 2p, or adsorbate orbitals), or state-averaging details. This directly affects the claimed ordering and persistence of S1/T2 states that underpin the 257 nm vs 343 nm photochemical distinction in the abstract.
- [Results and Discussion] Results/Discussion (barrier comparisons): No convergence criteria, basis-set details, slab thickness, or k-point sampling are reported for the PBE-D3 optimizations. Because PBE is known to delocalize electrons on reduced TiO2 and underestimate gaps, even modest shifts in PCET vs HAT barriers could reverse the claimed dominance on stoichiometric versus oxidized surfaces.
- [Abstract / Photochemical Pathways] Abstract and photochemical section: The assertion that 257 nm excitation allows the system to 'persist in higher excited states (S1/T2)' and bypass the ground-state barrier lacks supporting non-adiabatic dynamics, state lifetimes, or oscillator-strength data. Without these, the wavelength-dependent mechanism remains an interpretation rather than a demonstrated outcome.
minor comments (3)
- [Abstract] The abstract states that oxidation causes a 'significant increase in styrene yield' but supplies neither the numerical factor nor a direct citation to the experimental data being interpreted.
- [Figures] Energy diagrams and potential-energy surfaces should explicitly label the level of theory (PBE-D3 or SA-CASSCF) and indicate whether zero-point corrections or dispersion are included.
- [Discussion] A short table comparing computed barriers to any available experimental activation energies or prior DFT studies on similar TiO2 systems would strengthen the mechanistic claims.
Simulated Author's Rebuttal
We are grateful to the referee for the detailed and insightful comments, which have helped us identify areas for improvement in clarity and completeness. Below, we provide point-by-point responses to the major comments and outline the revisions we intend to implement.
read point-by-point responses
-
Referee: [Computational Methods] Computational Methods (or equivalent section): The SA-CASSCF calculations are described without specifying the active-space size, orbital selection (e.g., inclusion of Ti 3d, O 2p, or adsorbate orbitals), or state-averaging details. This directly affects the claimed ordering and persistence of S1/T2 states that underpin the 257 nm vs 343 nm photochemical distinction in the abstract.
Authors: We appreciate the referee's observation. The details of the SA-CASSCF setup were omitted in the submitted manuscript. In the revised version, we will add a comprehensive description of the active space (including the number of electrons and orbitals, with selection based on Ti 3d, O 2p, and adsorbate contributions), orbital selection procedure, and state-averaging details to ensure reproducibility and to support the excited-state analysis. revision: yes
-
Referee: [Results and Discussion] Results/Discussion (barrier comparisons): No convergence criteria, basis-set details, slab thickness, or k-point sampling are reported for the PBE-D3 optimizations. Because PBE is known to delocalize electrons on reduced TiO2 and underestimate gaps, even modest shifts in PCET vs HAT barriers could reverse the claimed dominance on stoichiometric versus oxidized surfaces.
Authors: We agree that these parameters are crucial for assessing the reliability of the calculations. We will include all missing technical details in the Computational Methods section of the revised manuscript, including convergence thresholds, basis sets, slab model specifications, and k-point grids. Additionally, we will add a brief discussion addressing the known limitations of PBE for TiO2 systems and argue why the relative barrier comparisons remain valid for distinguishing PCET and HAT mechanisms. revision: yes
-
Referee: [Abstract / Photochemical Pathways] Abstract and photochemical section: The assertion that 257 nm excitation allows the system to 'persist in higher excited states (S1/T2)' and bypass the ground-state barrier lacks supporting non-adiabatic dynamics, state lifetimes, or oscillator-strength data. Without these, the wavelength-dependent mechanism remains an interpretation rather than a demonstrated outcome.
Authors: We concur that the photochemical pathway description relies on an interpretation of the static electronic structure calculations rather than dynamical evidence. The manuscript does not include non-adiabatic molecular dynamics, lifetime estimates, or oscillator strengths. In the revision, we will rephrase the abstract and the photochemical pathways discussion to clearly indicate that the proposed persistence in S1/T2 states at 257 nm (versus relaxation at 343 nm) is deduced from the computed state energies, characters, and comparison to ground-state barriers. This will temper the claim appropriately while retaining the mechanistic insight. revision: yes
- Providing explicit non-adiabatic dynamics simulations, state lifetimes, or oscillator strength data to directly validate the wavelength-dependent excited-state persistence, as these were not part of the performed calculations.
Circularity Check
No circularity: standard quantum-chemistry results on defined models
full rationale
The paper reports DFT-PBE-D3 geometry optimizations and SA-CASSCF excited-state calculations for ethylbenzene dehydrogenation pathways on stoichiometric and oxidized TiO2(110) models. All mechanistic claims (PCET dominance, wavelength-dependent barrier bypass, HAT on oxidized surfaces) are direct outputs of these electronic-structure computations rather than derivations, fitted parameters renamed as predictions, or self-citation chains. No equations reduce to their inputs by construction, no uniqueness theorems are imported from prior author work, and no ansatz is smuggled via citation. The work is self-contained against external benchmarks (computed barriers, state characters) and contains no load-bearing self-referential steps.
Axiom & Free-Parameter Ledger
free parameters (2)
- PBE-D3 dispersion parameters
- SA-CASSCF active space
axioms (2)
- standard math Born-Oppenheimer approximation is valid for separating nuclear and electronic motion in the surface reaction.
- domain assumption The periodic slab model of rutile TiO2(110) sufficiently represents experimental surface conditions.
Reference graph
Works this paper leans on
- [1]
-
[2]
I. G. Ryabinkin, L. Joubert-Doriol, A. F. Izmaylov:Acc. Chem. Res.(2017),50, 1785–1793
work page 2017
-
[3]
J. P. Malhado, M. J. Bearpark, J. T. Hynes:Front. Chem.(2014),2, 1–21
work page 2014
-
[4]
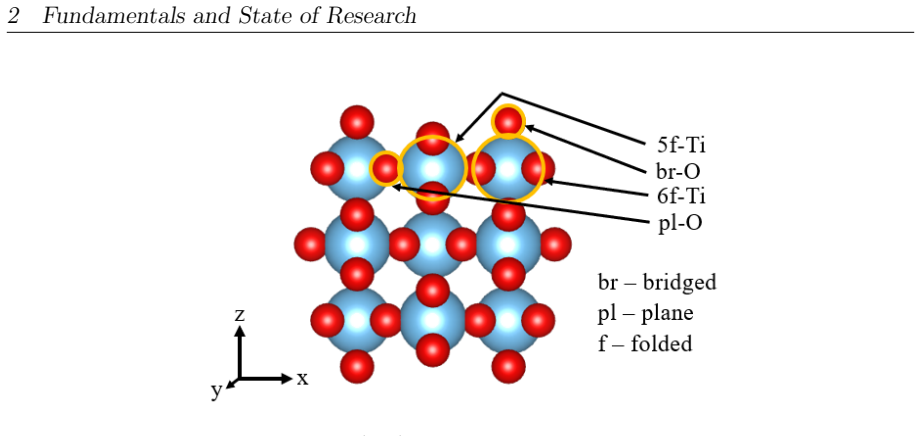
Y. Chen, D. Dang, B. Yan, Y. Cheng:Catalysts(2022),12, 71. 74 List of Abbreviations 5f-Ti - Five-fold coordinated Titanium 6f-Ti - Six-fold coordinated Titanium ABS - Acrylonitrile Butadiene Styrene AO - Atomic Orbitals ASCT - Adsorbate-to-Surface Charge Transfer BR - Boundary Region BSIE - Basis Set Incompleteness Error BSSE - Basis Set Superposition Err...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.