scHelix: Asymmetric Dual-Stream Integration via Explicit Gene-Level Disentanglement
Pith reviewed 2026-05-20 12:50 UTC · model grok-4.3
The pith
scHelix integrates scRNA-seq data by partitioning genes into stable anchors and variable variants then aligning streams asymmetrically to remove batch effects while preserving biology.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
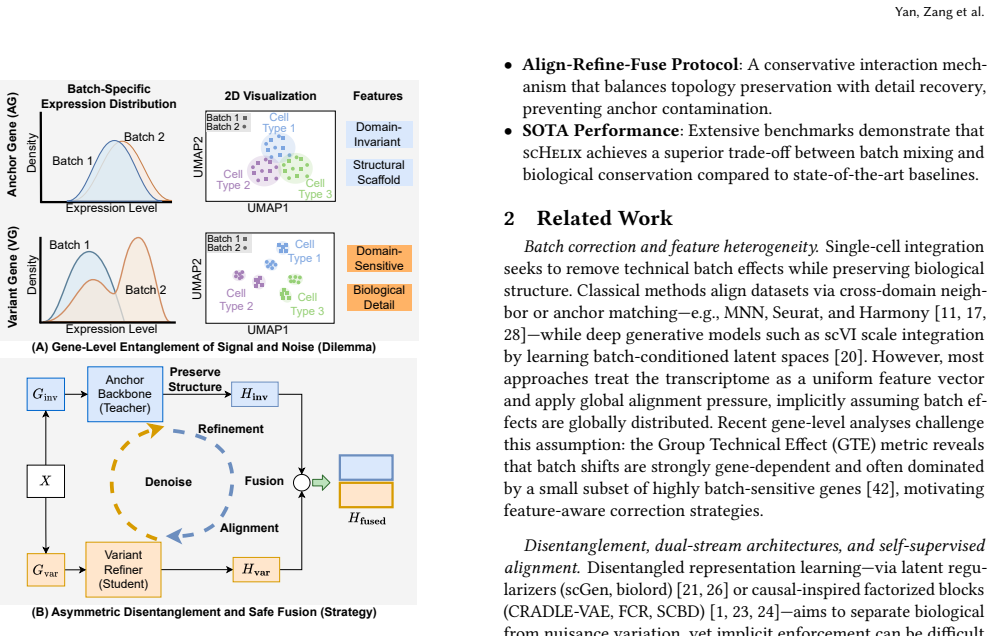
scHelix partitions the transcriptome into domain-invariant Anchor genes and domain-sensitive Variant genes before any model processing. It employs a dual-stream sparse diffusion encoder with stop-gradient graph caching to capture multi-scale structure. The central mechanism is an asymmetric Align-Refine-Fuse protocol that first aligns the unstable Variant stream to the robust topology of the Anchor stream and then lets the Anchor stream absorb denoised details from the Variant stream via bounded residual gating. This divide-and-conquer design prevents shortcut learning and delivers robust batch removal without loss of biological cluster integrity.
What carries the argument
asymmetric Align-Refine-Fuse protocol that aligns the Variant stream to the Anchor stream's topology and then refines the Anchor stream with bounded residual gating
If this is right
- Batch effects can be removed more effectively once genes are pre-partitioned by their sensitivity to technical variation.
- The dual-stream architecture with asymmetric alignment prevents the model from learning shortcuts that mix technical and biological signals.
- Subtle biological differences remain visible because the stable Anchor stream supplies a fixed reference topology for the alignment step.
- Overall performance on standard integration benchmarks improves relative to methods that treat every gene the same way.
Where Pith is reading between the lines
- The same anchor-variant split could be tested on other single-cell data types such as chromatin accessibility or spatial transcriptomics where batch variation is also feature-specific.
- Replacing the fixed pre-partitioning step with a learned gating module might allow the model to adapt the split during training on new datasets.
- The bounded residual gating idea offers a general way to control information exchange between noisy and clean streams in other multi-modal biological models.
Load-bearing premise
Batch effects appear differently across individual genes, so genes can be correctly separated into domain-invariant anchors and domain-sensitive variants right at the input before any learning occurs.
What would settle it
Construct a synthetic dataset in which the same batch shift is added uniformly to every gene and check whether scHelix still outperforms standard uniform-processing integration methods while keeping biological clusters intact.
Figures







read the original abstract
A critical challenge in single-cell RNA sequencing (scRNA-seq) integration is resolving the tension between eliminating batch effects and maintaining biological fidelity. While recent evidence indicates that batch effects manifest heterogeneously across genes, most existing methods process the transcriptome uniformly, frequently resulting in over-correction and loss of subtle biological signals. To address this, we present scHelix, a dataset-adaptive framework that fundamentally changes how features are processed by explicitly partitioning genes into domain-invariant Anchors and domain-sensitive Variants at the input level. scHelix utilizes a dual-stream sparse diffusion encoder equipped with stop-gradient graph caching to efficiently learn multi-scale structural representations. The core of our approach is a novel asymmetric Align-Refine-Fuse protocol: the unstable Variant stream is first aligned to the robust topology of the Anchor stream, followed by a conservative refinement phase where the Anchor stream absorbs denoised details via bounded residual gating. This divide-and-conquer architecture prevents shortcut learning and ensures robust batch removal without compromising the integrity of biological clusters. Extensive benchmarking demonstrates that scHelix outperforms state-of-the-art methods.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces scHelix, a dataset-adaptive framework for scRNA-seq batch integration. It explicitly partitions genes at the input level into domain-invariant Anchors and domain-sensitive Variants, processes them via a dual-stream sparse diffusion encoder with stop-gradient graph caching, and applies an asymmetric Align-Refine-Fuse protocol in which the Variant stream is aligned to the Anchor stream followed by bounded residual gating refinement. The central claim is that this divide-and-conquer design prevents shortcut learning and achieves robust batch removal without compromising biological cluster integrity, with extensive benchmarking asserted to show outperformance over state-of-the-art methods.
Significance. If the input-level partitioning proves reliable and the empirical results hold, the approach could meaningfully advance single-cell integration by explicitly addressing heterogeneous batch effects across genes rather than applying uniform processing, potentially reducing over-correction while preserving subtle biological signals. The asymmetric protocol and diffusion-based structural representations constitute a distinct architectural choice that, if substantiated, would differentiate it from existing uniform or post-hoc correction pipelines.
major comments (2)
- [Abstract] Abstract: The assertion that 'extensive benchmarking demonstrates that scHelix outperforms state-of-the-art methods' is unsupported by any quantitative results, error bars, dataset details, ablation studies, or performance tables in the manuscript. This absence renders the central performance claim unverifiable and load-bearing for the contribution.
- [Method] Method (partitioning description): The framework depends on an explicit, unsupervised input-level partition of genes into Anchors and Variants whose definition, statistical heuristic, or algorithmic procedure is not specified. Because this separation is presented as the prerequisite that enables the asymmetric protocol to avoid shortcut learning, the lack of detail makes it impossible to assess whether the claimed robustness follows independently or reduces to a re-expression of the input selection rule.
minor comments (1)
- [Abstract] The abstract introduces 'stop-gradient graph caching' and 'bounded residual gating' without defining their precise implementation or providing pseudocode, which hinders reproducibility even if the high-level protocol is sound.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback. We address each major comment below and indicate the revisions we will make to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: The assertion that 'extensive benchmarking demonstrates that scHelix outperforms state-of-the-art methods' is unsupported by any quantitative results, error bars, dataset details, ablation studies, or performance tables in the manuscript. This absence renders the central performance claim unverifiable and load-bearing for the contribution.
Authors: We agree that the abstract's performance claim would benefit from greater specificity to allow immediate verification. Although the full manuscript contains an Experiments section with benchmarking across multiple datasets, performance tables, and ablation studies, we will revise the abstract to include a concise summary of key quantitative outcomes (e.g., average improvements in ARI and batch-effect metrics with reference to the number of datasets and runs). This change will make the central claim self-contained while preserving the abstract's brevity. revision: yes
-
Referee: [Method] Method (partitioning description): The framework depends on an explicit, unsupervised input-level partition of genes into Anchors and Variants whose definition, statistical heuristic, or algorithmic procedure is not specified. Because this separation is presented as the prerequisite that enables the asymmetric protocol to avoid shortcut learning, the lack of detail makes it impossible to assess whether the claimed robustness follows independently or reduces to a re-expression of the input selection rule.
Authors: We acknowledge that the current manuscript does not provide an explicit description of the unsupervised procedure used to partition genes into Anchors and Variants. This detail is essential for evaluating the contribution. In the revised version we will add a dedicated subsection in Methods that specifies the statistical heuristic, including the exact criteria and algorithmic steps for the input-level separation. This addition will clarify that the partition is performed independently of the downstream asymmetric protocol. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper presents explicit input-level partitioning of genes into Anchors and Variants as a foundational, dataset-adaptive step justified by external recent evidence on heterogeneous batch effects. The asymmetric Align-Refine-Fuse protocol and dual-stream encoder are then described as building upon this partition to achieve the claimed robustness. No equations, fitted parameters, or self-citations are exhibited that reduce the core claims or predictions back to the inputs by construction. The derivation chain remains self-contained with independent architectural content.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Batch effects manifest heterogeneously across genes
invented entities (1)
-
domain-invariant Anchors and domain-sensitive Variants
no independent evidence
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
explicitly partitioning genes into domain-invariant Anchors and domain-sensitive Variants at the input level... asymmetric Align-Refine-Fuse protocol
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Seungheun Baek, Soyon Park, Yan Ting Chok, Junhyun Lee, Jueon Park, Mogan Gim, and Jaewoo Kang. 2024. CRADLE-VAE: Enhancing Single-Cell Gene Pertur- bation Modeling with Counterfactual Reasoning-based Artifact Disentanglement. arXiv:2409.05484 [cs.LG] doi:10.48550/arXiv.2409.05484 arXiv v2 (last revised 2024-09-10)
-
[2]
Seungbyn Baek, Kyungwoo Song, and Insuk Lee. 2025. Single-cell foundation models: bringing artificial intelligence into cell biology.Experimental & Molecular Medicine57 (2025), 2169–2181. doi:10.1038/s12276-025-01547-5
-
[4]
Xinlei Chen and Kaiming He. 2021. Exploring Simple Siamese Representation Learning. InProceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR). IEEE, Piscataway, NJ, USA, 11 pages
work page 2021
-
[5]
WebProtÃľgÃľ: a collaborative Web-based platform for editing biomedical ontologies
Sriram P. Chockalingam, Maneesha Aluru, and Srinivas Aluru. 2025. SCE- MENT: scalable and memory efficient integration of large-scale single-cell RNA- sequencing data.Bioinformatics41, 2 (2025), btaf057. doi:10.1093/bioinformatics/ btaf057
-
[6]
Haotian Cui, Cheng Wang, Hammad Maan, Kevin Pang, Feng Luo, Nan Duan, and Bo Wang. 2024. scGPT: toward building a foundation model for single- cell multi-omics using generative AI.Nature Methods21, 8 (2024), 1470–1480. doi:10.1038/s41592-024-02201-0
-
[7]
Mingming Gong, Kun Zhang, Tongliang Liu, Dacheng Tao, Clark Glymour, and Bernhard Schölkopf. 2016. Domain Adaptation with Conditional Transferable Components. InProceedings of The 33rd International Conference on Machine Learning (Proceedings of Machine Learning Research, Vol. 48), Maria Florina Balcan and Kilian Q. Weinberger (Eds.). PMLR, New York, New ...
work page 2016
-
[9]
Jean-Bastien Grill, Florian Strub, Florent Altché, et al. 2020. Bootstrap Your Own Latent: A New Approach to Self-Supervised Learning. InAdvances in Neural Information Processing Systems (NeurIPS). Curran Associates, Inc., Red Hook, NY, USA, 21271–21284
work page 2020
-
[10]
David Ha, Andrew Dai, and Quoc V. Le. 2017. HyperNetworks. InInternational Conference on Learning Representations (ICLR). OpenReview.net, Toulon, France, 18 pages
work page 2017
-
[11]
Laleh Haghverdi, Aaron T. L. Lun, Michael D. Morgan, and John C. Marioni
-
[12]
Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors.Nature Biotechnology36, 5 (2018), 421–427. doi:10. 1038/nbt.4091
work page 2018
-
[13]
Geoffrey Hinton, Oriol Vinyals, and Jeff Dean. 2015. Distilling the Knowledge in a Neural Network. arXiv preprint arXiv:1503.02531. Presented at NeurIPS 2015 Deep Learning Workshop
work page internal anchor Pith review Pith/arXiv arXiv 2015
-
[14]
Karin Hrovatin, Amir Ali Moinfar, Luke Zappia, Shrey Parikh, Alejandro Te- jada Lapuerta, Ben Lengerich, Manolis Kellis, and Fabian J. Theis. 2025. Integrat- ing single-cell RNA-seq datasets with substantial batch effects.BMC Genomics 26, 1 (2025), 974. doi:10.1186/s12864-025-12126-3
-
[15]
Johannes Klicpera, Aleksandar Bojchevski, and Stephan Günnemann. 2019. Pre- dict then Propagate: Graph Neural Networks meet Personalized PageRank. In International Conference on Learning Representations (ICLR). OpenReview.net, New Orleans, LA, USA, 15 pages
work page 2019
-
[16]
Andrew L Koenig, Irina Shchukina, Junedh Amrute, Patrick S Anber, Katarina Yaber, Kory J Lavine, Sanjoy Bhattacharya, Kamran A Bhojani, Jesse Koenig, et al. 2022. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure.Nature Cardiovascular Research1, 3 (2022), 263–280. doi:10.1038/s44161-022-00028-6
-
[19]
Ilya Korsunsky, Nghia Millard, Jean Fan, Kamil Slowikowski, Fan Zhang, Kevin Wei, Yuriy Baglaenko, Michael Brenner, Po-Ru Loh, and Soumya Raychaudhuri
-
[20]
Nature Methods16, 12 (2019), 1289–1296
Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods16, 12 (2019), 1289–1296. doi:10.1038/s41592-019-0619-0
-
[21]
Jeffrey T. Leek, Robert B. Scharpf, Héctor Corrada Bravo, David Simcha, Benjamin Langmead, W. Evan Johnson, Donald Geman, Keith Baggerly, and Rafael A. Irizarry. 2010. Tackling the widespread and critical impact of batch effects in high-throughput data.Nature Reviews Genetics11, 10 (2010), 733–739. doi:10. 1038/nrg2825
work page 2010
-
[22]
Haiping Liu, Shaojie Zhang, Shengzhong Mao, Qian Zhao, Yuxi Zhou, Andrew P. Gilmore, Mauricio A. Alvarez, and Hongpeng Zhou. 2025. BioBatchNet: A Dual-Encoder Framework for Robust Batch Effect Correction in Imaging Mass Cytometry. bioRxiv preprint. doi:10.1101/2025.03.15.643447
-
[23]
Romain Lopez, Jeffrey Regier, Michael B. Cole, Michael I. Jordan, and Nir Yosef
-
[24]
Deep generative modeling for single-cell transcriptomics.Nature Methods 15, 12 (2018), 1053–1058. doi:10.1038/s41592-018-0229-2
-
[25]
Mohammad Lotfollahi, F. Alexander Wolf, and Fabian J. Theis. 2019. scGen predicts single-cell perturbation responses.Nature Methods16, 8 (2019), 715–721. doi:10.1038/s41592-019-0494-8
-
[26]
Malte D. Luecken, M. Büttner, K. Chaichoompu, A. Danese, M. Interlandi, M. F. Mueller, D. C. Strobl, L. Zappia, M. Dugas, M. Colomé-Tatché, and Fabian J. Theis
-
[27]
doi:10.1038/s41592-021-01336-8
Benchmarking atlas-level data integration in single-cell genomics.Nature Methods19, 1 (2022), 41–50. doi:10.1038/s41592-021-01336-8
-
[28]
Taro Makino, Ji Won Park, Natasa Tagasovska, Takamasa Kudo, Paula Coelho, Jan-Christian Huetter, Heming Yao, Burkhard Hoeckendorf, Ana Carolina Leote, Stephen Ra, David Richmond, Kyunghyun Cho, Aviv Regev, and Romain Lopez
-
[29]
arXiv:2502.07281 [cs.LG] doi:10.48550/arXiv.2502.07281 arXiv v1 (submitted 2025-02-11)
Supervised Contrastive Block Disentanglement. arXiv:2502.07281 [cs.LG] doi:10.48550/arXiv.2502.07281 arXiv v1 (submitted 2025-02-11)
-
[30]
Haiyi Mao, Romain Lopez, Kai Liu, Jan-Christian Huetter, David Richmond, Panayiotis V. Benos, and Lin Qiu. 2024. Learning Identifiable Factorized Causal Representations of Cellular Responses. arXiv:2410.22472 [cs.LG] doi:10.48550/ arXiv.2410.22472 arXiv v3 (last revised 2024-12-02)
-
[31]
Mauro J Muraro, Gitanjali Dharmadhikari, Dominic Grün, Nathalie Groen, Tim Dielen, Erik Jansen, Leon Van Gurp, Marten A Engelse, Francoise Carlotti, Eelco Jp De Koning, et al . 2016. A single-cell transcriptome atlas of the hu- man pancreas.Cell systems3, 4 (2016), 385–394
work page 2016
-
[32]
Zohar Piran, Noga Cohen, Yedid Hoshen, and Mor Nitzan. 2024. Disentanglement of single-cell data with biolord.Nature Biotechnology42 (2024), 766–774. doi:10. 1038/s41587-023-01927-2
work page 2024
-
[33]
Simon, Yin-Ying Wang, and Zhongming Zhao
Lukas M. Simon, Yin-Ying Wang, and Zhongming Zhao. 2021. Integration of millions of transcriptomes using batch-aware triplet neural networks.Nature Machine Intelligence3 (2021), 705–715. doi:10.1038/s42256-021-00361-8
-
[34]
Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija
Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. 2019. Comprehensive integration of single-cell data.Cell177, 7 (2019), 1888–1902.e21. doi:10.1016/j.cell.2019.05.031
-
[35]
Antti Tarvainen and Harri Valpola. 2017. Mean teachers are better role models: Weight-averaged consistency targets improve semi-supervised deep learning results. InAdvances in Neural Information Processing Systems (NeurIPS). Curran Associates, Inc., Red Hook, NY, USA, 1195–1204
work page 2017
-
[36]
The Tabula Muris Consortium. 2020. A single-cell transcriptomic atlas char- acterizes ageing tissues in the mouse.Nature583, 7817 (2020), 590–595. doi:10.1038/s41586-020-2496-1
-
[37]
Theodoris, Ling Xiao, Akash Chopra, et al
Christina V. Theodoris, Ling Xiao, Akash Chopra, et al. 2023. Transfer learning enables predictions in network biology.Nature618, 7965 (2023), 616–624. doi:10. 1038/s41586-023-06139-9
work page 2023
-
[38]
Traag, Ludo Waltman, and Nees Jan Van Eck
Vincent A. Traag, Ludo Waltman, and Nees Jan Van Eck. 2019. From Louvain to Leiden: guaranteeing well-connected communities.Scientific Reports9, 1 (2019),
work page 2019
-
[39]
doi:10.1038/s41598-019-41695-z
-
[40]
Cole Trapnell. 2015. Defining cell types and states with single-cell genomics. Genome Research25, 10 (2015), 1491–1498. doi:10.1101/gr.190595.115
-
[41]
Kyle J Travaglini, Ahmad N Nabhan, Lolita Penland, Rahul Sinha, Astrid Gillich, Rene V Sit, Stephen Chang, Stephanie D Conley, Yelena Mber, Mia Huff, et al
-
[42]
Nature587, 7835 (2020), 619–625
A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature587, 7835 (2020), 619–625. doi:10.1038/s41586-020-2922-4
-
[43]
Gomez, Łukasz Kaiser, and Illia Polosukhin
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N. Gomez, Łukasz Kaiser, and Illia Polosukhin. 2017. Attention is All you Need. InAdvances in Neural Information Processing Systems (NeurIPS). Cur- ran Associates, Inc., Red Hook, NY, USA, 5998–6008
work page 2017
-
[44]
Juexin Wang, Anjun Ma, Yuzhou Chang, Jianting Gong, Yuexu Jiang, Ren Qi, Cankun Wang, Hongjun Fu, Qin Ma, and Dong Xu. 2021. scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses.Nature Communications12, 1 (2021), 1882. doi:10.1038/s41467-021-22197-x
-
[45]
Alexander Wolf, Philipp Angerer, and Fabian J
F. Alexander Wolf, Philipp Angerer, and Fabian J. Theis. 2018. SCANPY: large- scale single-cell gene expression data analysis.Genome Biology19, 1 (2018), 15. doi:10.1186/s13059-017-1382-0
-
[46]
Lei Xiong, Kang Tian, Yuzhe Li, Weixi Ning, Xin Gao, and Qiangfeng Cliff Zhang
-
[47]
Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space.Nature Communications13 (2022),
work page 2022
-
[48]
doi:10.1038/s41467-022-33758-z
-
[49]
Chenling Xu, Romain Lopez, Edouard Mehlman, Jeffrey Regier, Michael I. Jordan, and Nir Yosef. 2021. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models.Molecular Systems Biology17, 1 (2021), e9620. doi:10.15252/msb.20209620
-
[50]
Bowen Zhao, Kailu Song, Dong-Qing Wei, Yi Xiong, and Jun Ding. 2025. scCobra allows contrastive cell embedding learning with domain adaptation for single Yan, Zang et al. cell data integration and harmonization.Communications Biology8 (2025), 233. doi:10.1038/s42003-025-07692-x
-
[52]
Yang Zhou, Qiongyu Sheng, Guohua Wang, Li Xu, and Shuilin Jin. 2025. Quanti- fying batch effects for individual genes in single-cell data.Nature Computational Science5, 8 (2025), 612–620. doi:10.1038/s43588-025-00824-7 A Theoretical Analysis In this section, we provide rigorous theoretical justifications for the “divide-and-conquer” mechanisms inscHelix. ...
-
[53]
For any inputv∈R 𝑑, LN(v)= v−𝜇 𝜎 ⊙𝜸+𝜷
Global Boundedness (Safety Valve).The term Δh𝑖 is the output of a LayerNorm (LN) operation. For any inputv∈R 𝑑, LN(v)= v−𝜇 𝜎 ⊙𝜸+𝜷. Since the normalized term (v−𝜇)/𝜎 has unit variance, its Euclidean norm is exactly √ 𝑑. Thus, ∥LN( v) ∥2 ≤ √ 𝑑∥𝜸∥ ∞ + ∥𝜷∥ 2 =: 𝐶LN. Combined with the sigmoid gate ∥𝜶𝑖 ∥∞ ≤𝛼 max, the total perturba- tion is bounded: ∥ ˜Hinv 𝑖 −...
-
[54]
Lipschitz Continuity (Smoothness).The functions compos- ing the update—Sigmoid, LayerNorm, and Softmax (in Attention)— are all Lipschitz continuous and differentiable almost everywhere. This implies there exists a constant𝐿such that: ∥Δ ˜hinv 𝑖 (x) −Δ ˜hinv 𝑖 (y) ∥2 ≤𝐿∥x−y∥ 2 (18) Yan, Zang et al. for any two Variant inputsx , y. Consequently, small pertu...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.