Formal O(N3)-Scaling Second-Order Perturbation Theory by Block Tensor Decomposition: Implementation on MP2 and rPT2
Pith reviewed 2026-06-29 09:19 UTC · model grok-4.3
The pith
Block tensor decomposition reduces second-order perturbation theory to formal O(N^3) scaling while matching RI-MP2 accuracy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
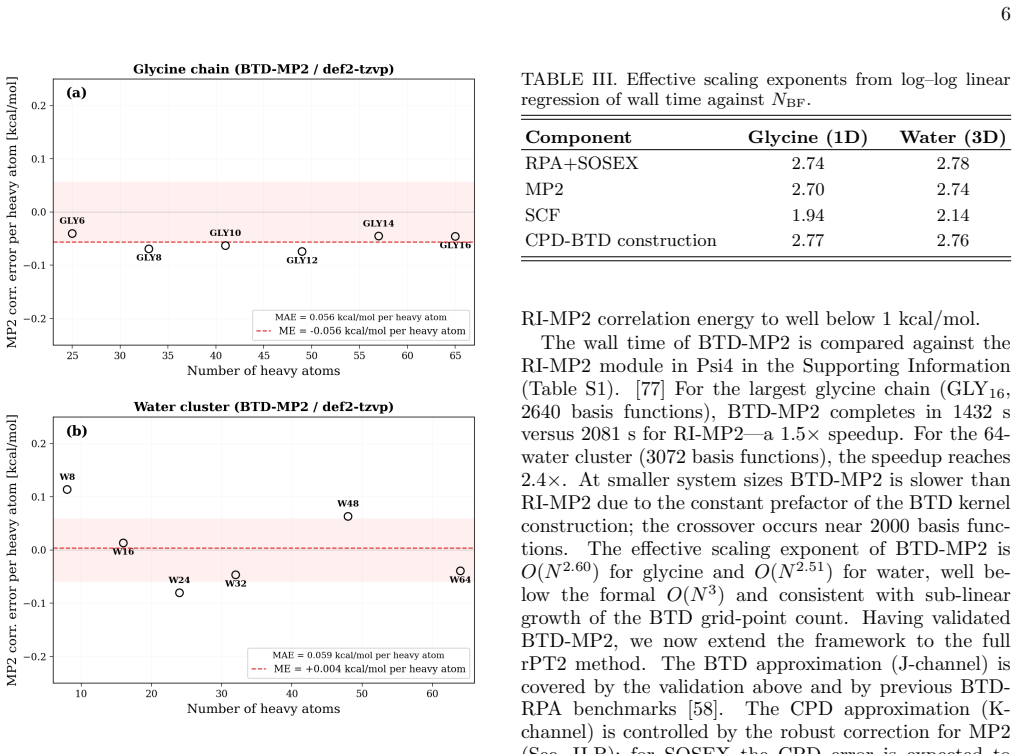
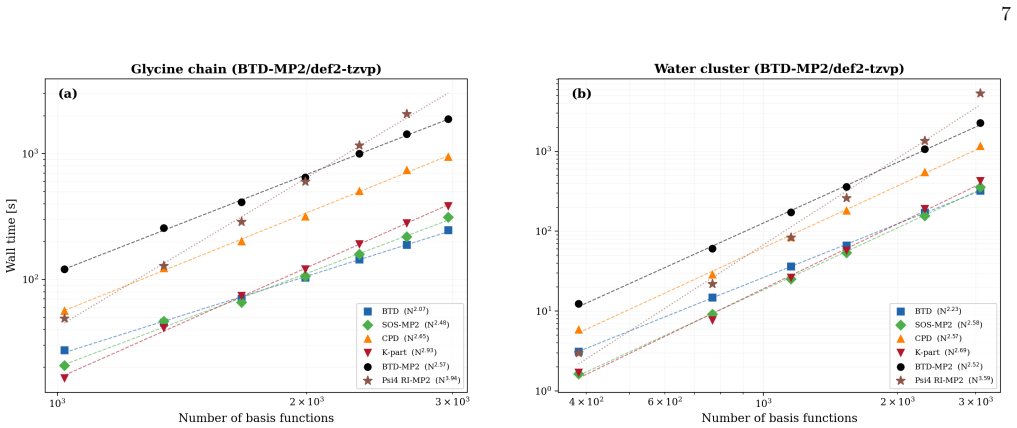
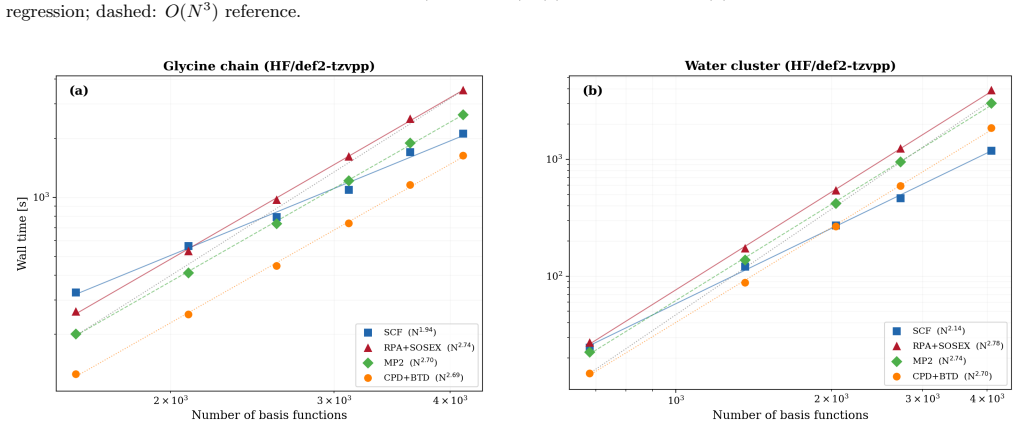
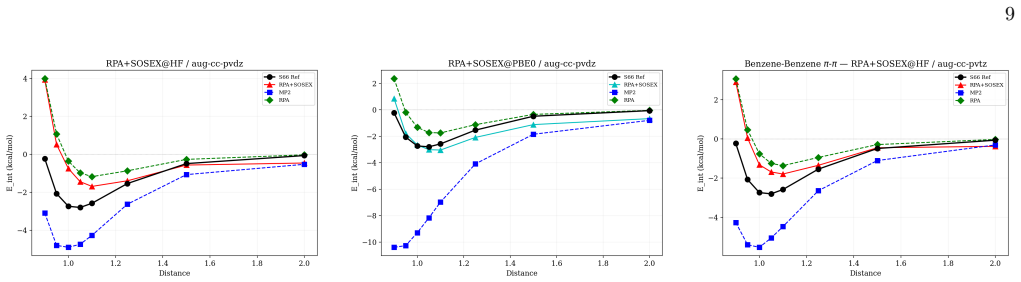
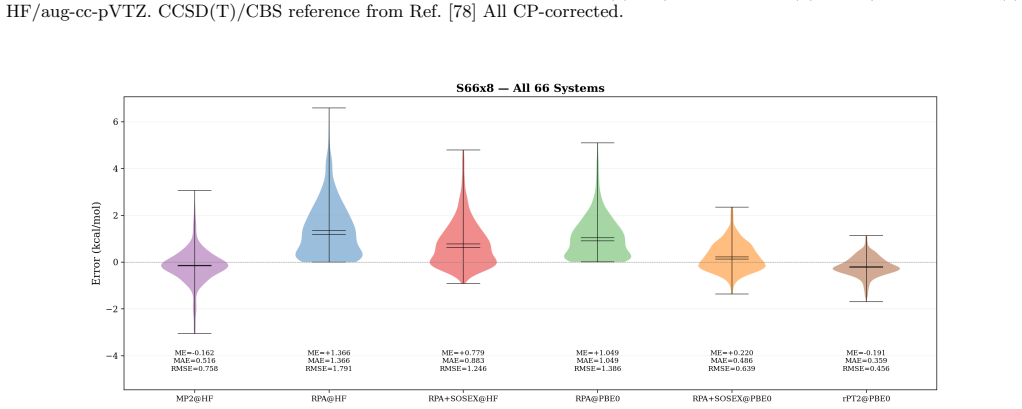
Block tensor decomposition constructs the tensor hyper-contraction kernel at O(N^3) via a dual-grid scheme; canonical polyadic decomposition factorizes the exchange channel through a block-based two-stage ALS. An asymmetric half-kernel design applies bare Coulomb to one vertex and coupling-constant-averaged screening to the other, capturing the SOSEX component of rPT2 without a frequency-dependent CPD. For MP2 this reproduces canonical RI-MP2 to 0.058 kcal/mol per heavy atom; for rPT2@PBE0 on the S66x8 benchmark the mean absolute error is 0.36 kcal/mol over 528 data points, with O(N^2) storage.
What carries the argument
The BTD-CPD framework, in which block tensor decomposition builds the hyper-contraction kernel at O(N^3) and canonical polyadic decomposition factorizes the exchange channel to deliver both the scaling and the accuracy.
If this is right
- MP2 and rPT2 calculations become feasible with cubic rather than higher scaling.
- Storage of intermediates drops from higher powers of N to quadratic.
- The SOSEX contribution in rPT2 is obtained without introducing frequency dependence into the CPD step.
- The same machinery applies uniformly to both MP2 and renormalized PT2 variants.
Where Pith is reading between the lines
- The approach could open PT2-level treatments to molecular systems previously limited by cost.
- The block decomposition pattern may transfer to other tensor contractions common in quantum chemistry.
- Direct comparison on systems with increasing numbers of heavy atoms would test whether the per-atom error remains bounded.
Load-bearing premise
The dual-grid BTD scheme and block-based CPD factorization must capture the tensor hyper-contraction kernel and exchange channel accurately for general molecules without errors that grow with system size or chemical complexity.
What would settle it
A BTD-CPD MP2 calculation on a molecule containing many heavy atoms that deviates from the canonical RI-MP2 result by substantially more than 0.058 kcal/mol per heavy atom would falsify the accuracy claim.
Figures
read the original abstract
Block tensor decomposition (BTD) and canonical polyadic decomposition (CPD) are combined into a unified $O(N^3)$-scaling framework for second-order perturbation theory (PT2), demonstrated on MP2 and renormalized PT2 (rPT2). BTD constructs the tensor hyper-contraction kernel at $O(N^3)$ via a dual-grid scheme; CPD factorizes the exchange channel through a block-based two-stage ALS. An asymmetric half-kernel design applies bare Coulomb to one vertex and coupling-constant-averaged screening to the other, capturing the SOSEX component of rPT2 without a frequency-dependent CPD. For MP2, BTD-CPD reproduces canonical RI-MP2 to 0.058~kcal/mol per heavy atom. For rPT2@PBE0 on the S66x8 benchmark, the mean absolute error is 0.36~kcal/mol (ME $-$0.19, RMSE 0.46) over 528 data points. The CPD-compressed intermediates yield $O(N^2)$ storage alongside $O(N^3)$ scaling.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a BTD-CPD framework that combines block tensor decomposition with a dual-grid scheme to construct the tensor hyper-contraction kernel at formal O(N^3) cost and block-based canonical polyadic decomposition (two-stage ALS) to factorize the exchange channel, applied to MP2 and rPT2. An asymmetric half-kernel design captures the SOSEX term in rPT2 without frequency-dependent factorization. On the S66x8 benchmark the method reproduces canonical RI-MP2 to 0.058 kcal/mol per heavy atom and yields 0.36 kcal/mol MAE (ME -0.19, RMSE 0.46) for rPT2@PBE0 over 528 points, with O(N^2) storage for the compressed intermediates.
Significance. If the formal O(N^3) scaling is rigorously established and the decomposition ranks remain bounded for general systems, the approach would enable correlated PT2 calculations on substantially larger molecules while preserving chemical accuracy and reducing storage, constituting a meaningful methodological advance in quantum chemistry.
major comments (1)
- [Benchmark results] Benchmark results section: the reported error bounds (0.058 kcal/mol per heavy atom for MP2; 0.36 kcal/mol MAE for rPT2) are obtained exclusively on the S66x8 set of small-to-medium organic complexes. No data or analysis is provided on how the required BTD grid density or CPD ranks scale with molecular size N or with increasing delocalization, which is necessary to confirm that the dual-grid BTD and block CPD errors remain bounded and do not compromise the formal O(N^3) scaling claim for arbitrary systems.
minor comments (2)
- [Introduction / Theory] The abstract and introduction should explicitly state the precise definition of the dual-grid BTD construction and the block partitioning used in the CPD factorization (e.g., which tensor blocks are treated independently) so that the O(N^3) scaling argument can be verified without ambiguity.
- Figure captions and table footnotes should clarify whether the quoted timings and storage figures include the cost of determining the decomposition ranks or are measured after rank selection.
Simulated Author's Rebuttal
We thank the referee for their constructive review and for highlighting the need to substantiate the scaling claims. We address the single major comment below.
read point-by-point responses
-
Referee: [Benchmark results] Benchmark results section: the reported error bounds (0.058 kcal/mol per heavy atom for MP2; 0.36 kcal/mol MAE for rPT2) are obtained exclusively on the S66x8 set of small-to-medium organic complexes. No data or analysis is provided on how the required BTD grid density or CPD ranks scale with molecular size N or with increasing delocalization, which is necessary to confirm that the dual-grid BTD and block CPD errors remain bounded and do not compromise the formal O(N^3) scaling claim for arbitrary systems.
Authors: We agree that the numerical benchmarks are confined to the S66x8 set. The formal O(N^3) scaling is obtained from the algorithmic construction: the dual-grid BTD evaluates the THC kernel in O(N^3) operations once the grid density is fixed, and the block CPD factorizes the exchange integrals via two-stage ALS whose leading cost is also O(N^3) for fixed rank. The manuscript demonstrates that, for the grid densities and ranks chosen on S66x8, the errors remain below chemical accuracy. We do not, however, supply explicit data showing how those ranks or grid densities must increase with N or with greater delocalization. This constitutes a genuine limitation of the current benchmark suite. We will revise the manuscript to (i) restate that the O(N^3) claim assumes decomposition parameters sufficient to reach the target accuracy and (ii) add an explicit discussion of the need for future studies on larger and more delocalized systems to verify that the required ranks remain moderate. revision: yes
Circularity Check
No significant circularity detected in the derivation
full rationale
The paper introduces a BTD-CPD framework for O(N^3) PT2, with explicit validation that BTD-CPD reproduces canonical RI-MP2 to 0.058 kcal/mol per heavy atom on S66x8 and reports independent MAE on rPT2 benchmarks. No quoted equations or steps reduce a claimed prediction to a fitted parameter by construction, nor does any load-bearing premise collapse to a self-citation chain or ansatz smuggled via prior work. The central scaling and accuracy claims rest on the tensor decomposition applied to the PT2 integrals, which are externally benchmarked rather than tautological.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Block tensor decomposition and canonical polyadic decomposition can be applied to the two-electron integrals and amplitudes of second-order perturbation theory without loss of formal scaling or essential accuracy.
Reference graph
Works this paper leans on
-
[1]
ˇC´ ıˇ zek, On the correlation problem in atomic and molecular systems
J. ˇC´ ıˇ zek, On the correlation problem in atomic and molecular systems. calculation of wavefunction compo- nents in Ursell-type expansion using quantum-field theo- retical methods, J. Chem. Phys.45, 4256 (1966)
1966
-
[2]
R. J. Bartlett, Many-body perturbation theory and cou- pled cluster theory for electron correlation in molecules, Annu. Rev. Phys. Chem.32, 359 (1981)
1981
-
[3]
R. J. Bartlett and M. Musia l, Coupled-cluster theory in quantum chemistry, Rev. Mod. Phys.79, 291 (2007)
2007
-
[4]
T. D. Crawford and H. F. Schaefer, An introduction to coupled cluster theory for computational chemists, Rev. Comput. Chem.14, 33 (2000)
2000
-
[5]
Shavitt and R
I. Shavitt and R. J. Bartlett,Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory(Cambridge University Press, Cambridge, 2009)
2009
-
[6]
Helgaker, P
T. Helgaker, P. Jørgensen, and J. Olsen,Molecular Electronic-Structure Theory(Wiley, Chichester, 2000)
2000
-
[7]
Møller and M
C. Møller and M. S. Plesset, Note on an approximation treatment for many-electron systems, Phys. Rev.46, 618 (1934)
1934
-
[8]
Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J
S. Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J. Chem. Phys. 124, 034108 (2006)
2006
-
[9]
Schwabe and S
T. Schwabe and S. Grimme, Double-hybrid density func- tionals with long-range dispersion corrections: Higher ac- curacy and extended applicability, Phys. Chem. Chem. Phys.9, 3397 (2007)
2007
-
[10]
Chai and M
J.-D. Chai and M. Head-Gordon, Long-range corrected double-hybrid density functionals, J. Chem. Phys.131, 174105 (2009)
2009
-
[11]
Zhang, X
Y. Zhang, X. Xu, and W. A. Goddard, Doubly hybrid density functional for accurate descriptions of nonbond interactions, thermochemistry, and thermochemical ki- netics, Proc. Natl. Acad. Sci. USA106, 4963 (2009)
2009
-
[12]
Goerigk and S
L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys.13, 6670 (2011)
2011
-
[13]
Goerigk and S
L. Goerigk and S. Grimme, Double-hybrid density func- tionals, WIREs Comput. Mol. Sci.4, 576 (2014)
2014
-
[14]
Mardirossian and M
N. Mardirossian and M. Head-Gordon, Survival of the most transferable at the top of Jacob’s ladder: Defining and testing theωB97M(2) double hybrid density func- tional, J. Chem. Phys.148, 241736 (2018)
2018
-
[15]
J. M. L. Martin and G. Santra, Empirical double-hybrid density functional theory: A ‘third way’ in between WFT and DFT, Isr. J. Chem.60, 787 (2020)
2020
-
[16]
Lee and M
J. Lee and M. Head-Gordon, Regularized orbital- optimized second-order Møller-Plesset perturbation the- ory: A reliable fifth-order-scaling electron correlation model with orbital energy dependent regularizers, J. Chem. Theory Comput.14, 5203 (2018)
2018
-
[17]
Carter-Fenk and M
K. Carter-Fenk and M. Head-Gordon, Repartitioned Brillouin-Wigner perturbation theory with a size- consistent second-order correlation energy, J. Chem. Phys.158, 234108 (2023)
2023
-
[18]
Carter-Fenk, J
K. Carter-Fenk, J. Shee, and M. Head-Gordon, Opti- mizing the regularization in size-consistent second-order Brillouin-Wigner perturbation theory, J. Chem. Phys. 159, 171104 (2023)
2023
-
[19]
Bohm and D
D. Bohm and D. Pines, A collective description of elec- tron interactions: III. Coulomb interactions in a degen- erate electron gas, Phys. Rev.92, 609 (1953)
1953
-
[20]
D. C. Langreth and J. P. Perdew, Exchange-correlation energy of a metallic surface: Wave-vector analysis, Phys. Rev. B15, 2884 (1977)
1977
-
[21]
Furche, Molecular tests of the random phase approx- imation to the exchange-correlation energy functional, Phys
F. Furche, Molecular tests of the random phase approx- imation to the exchange-correlation energy functional, Phys. Rev. B64, 195120 (2001)
2001
-
[22]
J. P. Perdew and K. Schmidt, Jacob’s ladder of density functional approximations for the exchange-correlation energy, AIP Conf. Proc.577, 1 (2001)
2001
-
[23]
Gunnarsson and B
O. Gunnarsson and B. I. Lundqvist, Exchange and corre- lation in atoms, molecules, and solids by the spin-density- functional formalism, Phys. Rev. B13, 4274 (1976)
1976
-
[24]
Eshuis, J
H. Eshuis, J. E. Bates, and F. Furche, Electron correla- tion methods based on the random phase approximation, Theor. Chem. Acc.131, 1084 (2012)
2012
-
[25]
X. Ren, P. Rinke, C. Joas, and M. Scheffler, Random- phase approximation and its applications in computa- tional chemistry and materials science, J. Mater. Sci.47, 7447 (2012)
2012
-
[26]
Gr¨ uneis, M
A. Gr¨ uneis, M. Marsman, J. Harl, L. Schimka, and G. Kresse, Making the random phase approximation to electronic correlation accurate, J. Chem. Phys.131, 154115 (2009)
2009
-
[27]
Paier, B
J. Paier, B. G. Janesko, T. M. Henderson, G. E. Scuse- ria, A. Gr¨ uneis, and G. Kresse, Hybrid functionals includ- ing random phase approximation correlation and second- order screened exchange, J. Chem. Phys.132, 094103 (2010)
2010
-
[28]
I. Y. Zhang and X. Xu, Simultaneous attenuation of both self-interaction error and nondynamic correlation error in density functional theory: A spin-pair distinc- tive adiabatic-connection approximation, J. Phys. Chem. Lett.10, 2617 (2019)
2019
-
[29]
Toulouse, I
J. Toulouse, I. C. Gerber, G. Jansen, A. Savin, and J. G. ´Angy´ an, Adiabatic-connection fluctuation- dissipation density-functional theory based on range sep- aration, Phys. Rev. Lett.102, 096404 (2009)
2009
-
[30]
D. L. Freeman, Coupled-cluster expansion applied to the electron gas: Inclusion of ring and exchange effects, Phys. Rev. B15, 5512 (1977)
1977
-
[31]
Jansen, R.-F
G. Jansen, R.-F. Liu, and J. G. ´Angy´ an, On the equiva- lence of ring-coupled-cluster doubles and adiabatic con- nection fluctuation-dissipation theorem random phase approximation, J. Chem. Phys.133, 154106 (2010)
2010
-
[32]
X. Ren, A. Tkatchenko, P. Rinke, and M. Scheffler, Be- yond the random-phase approximation for the electron correlation energy: The importance of single excitations, Phys. Rev. Lett.106, 153003 (2011)
2011
-
[33]
X. Ren, P. Rinke, G. E. Scuseria, and M. Scheffler, Renor- malized second-order perturbation theory for the elec- tron correlation energy: Concept, implementation, and benchmarks, Phys. Rev. B88, 035120 (2013)
2013
-
[34]
Neese, F
F. Neese, F. Wennmohs, and A. Hansen, Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method, J. Chem. Phys.130, 114108 (2009)
2009
-
[35]
Schmitz, C
G. Schmitz, C. H¨ attig, and D. P. Tew, Explicitly cor- related PNO-MP2-F12 and PNO-CCSD(F12*) methods, 11 Phys. Chem. Chem. Phys.16, 22167 (2014)
2014
-
[36]
Pinski, C
P. Pinski, C. Riplinger, E. F. Valeev, and F. Neese, Sparse maps—a systematic infrastructure for reduced- scaling electronic structure methods. I. An efficient and simple linear scaling local MP2 method, J. Chem. Phys. 143, 034108 (2015)
2015
-
[37]
Riplinger and F
C. Riplinger and F. Neese, An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys.138, 034106 (2013)
2013
-
[38]
Riplinger, B
C. Riplinger, B. Sandhoefer, A. Hansen, and F. Neese, Natural triple excitations in local coupled cluster calcu- lations with pair natural orbitals, J. Chem. Phys.139, 134101 (2013)
2013
-
[39]
Riplinger, P
C. Riplinger, P. Pinski, U. Becker, E. F. Valeev, and F. Neese, Sparse maps—a systematic infrastructure for reduced-scaling electronic structure methods. II. linear scaling domain based pair natural orbital coupled cluster theory, J. Chem. Phys.144, 024109 (2016)
2016
-
[40]
J. Yang, G. K.-L. Chan, F. R. Manby, M. Sch¨ utz, and H.-J. Werner, The orbital-specific-virtual local coupled cluster singles and doubles method, J. Chem. Phys.136, 144105 (2012)
2012
-
[41]
B. I. Dunlap, J. W. D. Connolly, and J. R. Sabin, On some approximations in applications of Xαtheory, J. Chem. Phys.71, 3396 (1979)
1979
-
[42]
Feyereisen, G
M. Feyereisen, G. Fitzgerald, and A. Komornicki, Use of approximate integrals in ab initio theory. An application in MP2 energy calculations, Chem. Phys. Lett.208, 359 (1993)
1993
-
[43]
N. H. F. Beebe and J. Linderberg, Simplifications in the generation and transformation of two-electron integrals in molecular calculations, Int. J. Quantum Chem.12, 683 (1977)
1977
-
[44]
Røeggen and T
I. Røeggen and T. Johansen, Cholesky decomposition of the two-electron integral matrix in electronic structure calculations, J. Chem. Phys.128, 194107 (2008)
2008
-
[45]
Neese, F
F. Neese, F. Wennmohs, A. Hansen, and U. Becker, Effi- cient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange, Chem. Phys.356, 98 (2009)
2009
-
[46]
R. A. Friesner, Solution of the Hartree–Fock equations by a pseudospectral method: Application to diatomic molecules, J. Chem. Phys.85, 1462 (1986)
1986
-
[47]
E. G. Hohenstein, R. M. Parrish, and T. J. Mart´ ınez, Tensor hypercontraction density fitting. I. Quartic scal- ing second- and third-order Møller-Plesset perturbation theory, J. Chem. Phys.137, 044103 (2012)
2012
-
[48]
R. M. Parrish, E. G. Hohenstein, T. J. Mart´ ınez, and C. D. Sherrill, Tensor hypercontraction. II. Least-squares renormalization, J. Chem. Phys.137, 224106 (2012)
2012
-
[49]
J. Lee, L. Lin, and M. Head-Gordon, Systematically im- provable tensor hypercontraction: Interpolative separa- ble density fitting for molecules applied to exact ex- change, second- and third-order Møller-Plesset pertur- bation theory, J. Chem. Theory Comput.16, 243 (2020)
2020
-
[50]
J. E. Moussa, Cubic-scaling algorithm and self-consistent field for the random-phase approximation with second- order screened exchange, J. Chem. Phys.140, 014107 (2014)
2014
-
[51]
Duchemin and X
I. Duchemin and X. Blase, Separable resolution-of-the- identity with all-electron Gaussian bases: Application to cubic-scaling RPA, J. Chem. Phys.150, 174120 (2019)
2019
-
[52]
Wilhelm, P
J. Wilhelm, P. Seewald, M. Del Ben, and J. Hutter, Large-scale cubic-scaling random phase approximation correlation energy calculations using a Gaussian basis, J. Chem. Theory Comput.12, 5851 (2016)
2016
-
[53]
H. F. Schurkus and C. Ochsenfeld, Communication: An effective linear-scaling atomic-orbital reformulation of the random-phase approximation using a contracted double-Laplace transformation, J. Chem. Phys.144, 031101 (2016)
2016
-
[54]
Yeh and M
C.-N. Yeh and M. Morales, Low-scaling algorithm for the random phase approximation using tensor hypercontrac- tion withk-point sampling, J. Chem. Theory Comput. 19, 6197 (2023)
2023
-
[55]
Yeh and M
C.-N. Yeh and M. Morales, Low-scaling algorithms for GWand constrained random phase approximation us- ing symmetry-adapted interpolative separable density fit- ting, J. Chem. Theory Comput.20, 3184 (2024)
2024
-
[56]
Zhang, W
Y. Zhang, W. Wu, and P. Su, FormalO(N 3) scaling GWcalculations by block tensor decomposition for large molecule systems, J. Chem. Phys.164, 144106 (2026)
2026
-
[57]
Y. Jung, R. C. Lochan, A. D. Dutoi, and M. Head- Gordon, Scaled opposite-spin second order Møller-Plesset correlation energy: An economical electronic structure method, J. Chem. Phys.121, 9793 (2004)
2004
-
[58]
Zhang, X
Y. Zhang, X. Xiong, W. Wu, and P. Su, Block tensor decomposition: A dual-grid scheme with a formalO(N 3) scale for THC decomposition of molecular systems, J. Chem. Phys.163, 174109 (2025)
2025
-
[59]
T. Y. Takeshita, W. A. de Jong, D. Neuhauser, R. Baer, and E. Rabani, Stochastic formulation of the resolution of identity: Application to second order Møller–Plesset perturbation theory, J. Chem. Theory Comput.13, 4605 (2017)
2017
-
[60]
W. Dou, M. Chen, T. Y. Takeshita, R. Baer, D. Neuhauser, and E. Rabani, Stochastic resolution of identity second-order Matsubara Green’s function the- ory, J. Chem. Phys.151, 044114 (2019)
2019
-
[61]
C. Zhao, Q. Ou, J. Lee, and W. Dou, Stochastic resolu- tion of identity to CC2 for large systems: Excited state properties, J. Chem. Theory Comput.20, 6211 (2024)
2024
-
[62]
F. L. Hitchcock, The expression of a tensor or a polyadic as a sum of products, J. Math. Phys.6, 164 (1927)
1927
-
[63]
Benedikt, A
U. Benedikt, A. A. Auer, M. Espig, and W. Hackbusch, Tensor decomposition in post-Hartree–Fock methods. I. Two-electron integrals and MP2, J. Chem. Phys.134, 054118 (2011)
2011
-
[64]
Pierce and M
K. Pierce and M. Morales, Using matrix-free tensor- network optimizations to construct a reduced-scaling and robust second-order Møller-Plesset theory, J. Chem. The- ory Comput.21, 5952 (2025)
2025
-
[65]
W. Hu, L. Lin, and C. Yang, Interpolative separable den- sity fitting decomposition for accelerating hybrid density functional calculations with applications to defects in sil- icon, J. Chem. Theory Comput.13, 5420 (2017)
2017
-
[66]
X. Qin, W. Hu, and J. Yang, Interpolative separable den- sity fitting for accelerating two-electron integrals: A the- oretical perspective, J. Chem. Theory Comput.19, 679 (2023)
2023
-
[67]
Alml¨ of, Elimination of energy denominators in Møller– Plesset perturbation theory by a Laplace transform ap- proach, Chem
J. Alml¨ of, Elimination of energy denominators in Møller– Plesset perturbation theory by a Laplace transform ap- proach, Chem. Phys. Lett.181, 319 (1991)
1991
-
[68]
H¨ aser and J
M. H¨ aser and J. Alml¨ of, Laplace transform techniques in Møller-Plesset perturbation theory, J. Chem. Phys.96, 489 (1992)
1992
-
[69]
Kaltak, J
M. Kaltak, J. Klimeˇ s, and G. Kresse, Low scaling al- gorithms for the random phase approximation: Imagi- 12 nary time and Laplace transformations, J. Chem. Theory Comput.10, 2498 (2014)
2014
-
[70]
Azizi, J
M. Azizi, J. Wilhelm, D. Golze, F. Delesma, R. Panad’es- Barrueta, P. Rinke, M. Giantomassi, and X. Gonze, Val- idation of the GreenX library time-frequency component for efficientGWand RPA calculations, Phys. Rev. B 109, 245101 (2024)
2024
-
[71]
X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter, and M. Scheffler, Resolution-of- identity approach to Hartree–Fock, hybrid density func- tionals, RPA, MP2 andGWwith numeric atom-centered orbital basis functions, New J. Phys.14, 053020 (2012)
2012
-
[72]
G. E. Scuseria, T. M. Henderson, and D. C. Sorensen, The ground state correlation energy of the random phase approximation from a ring coupled cluster doubles ap- proach, J. Chem. Phys.129, 231101 (2008)
2008
-
[73]
G. E. Scuseria, T. M. Henderson, and I. W. Bulik, Particle-particle and quasiparticle random phase approx- imations: Connections to coupled cluster theory, J. Chem. Phys.139, 104113 (2013)
2013
-
[74]
Bleiziffer, A
P. Bleiziffer, A. Heßelmann, and A. G¨ orling, Efficient self-consistent treatment of electron correlation within the random phase approximation, J. Chem. Phys.139, 084113 (2013)
2013
-
[75]
D. A. Matthews, Improved grid optimization and fitting in least squares tensor hypercontraction, J. Chem. The- ory Comput.16, 1382 (2020)
2020
-
[76]
Weigend, A fully direct RI-HF algorithm: Implemen- tation, optimised auxiliary basis sets, demonstration of accuracy and efficiency, Phys
F. Weigend, A fully direct RI-HF algorithm: Implemen- tation, optimised auxiliary basis sets, demonstration of accuracy and efficiency, Phys. Chem. Chem. Phys.4, 4285 (2002)
2002
-
[77]
Smith, L
D. Smith, L. Burns, A. Simmonett, R. Parrish, M. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Di Remi- gio, A. Alenaizan, A. James, S. Lehtola, J. Misiewicz, M. Scheurer, R. Shaw, J. Schriber, Y. Xie, Z. Glick, D. Sirianni, and C. Sherrill, Psi4 1.4: Open-source soft- ware for high-throughput quantum chemistry, J. Chem. Phys.152, 184108 (2020)
2020
-
[78]
ˇRez´ aˇ c, K
J. ˇRez´ aˇ c, K. E. Riley, and P. Hobza, S66: A Well- balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures, J. Chem. Theory Comput.7, 2427 (2011)
2011
-
[79]
S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate to- tal energies. Some procedures with reduced errors, Mol. Phys.19, 553 (1970)
1970
-
[80]
Jeziorski, R
B. Jeziorski, R. Moszynski, and K. Szalewicz, Perturba- tion theory approach to intermolecular potential energy surfaces of van der waals complexes, Chem. Rev.94, 1887 (1994)
1994
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.