GENEB: Why Genomic Models Are Hard to Compare
Pith reviewed 2026-06-28 06:21 UTC · model grok-4.3
The pith
GENEB shows that genomic model rankings vary sharply across task categories and architecture often outweighs scale.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
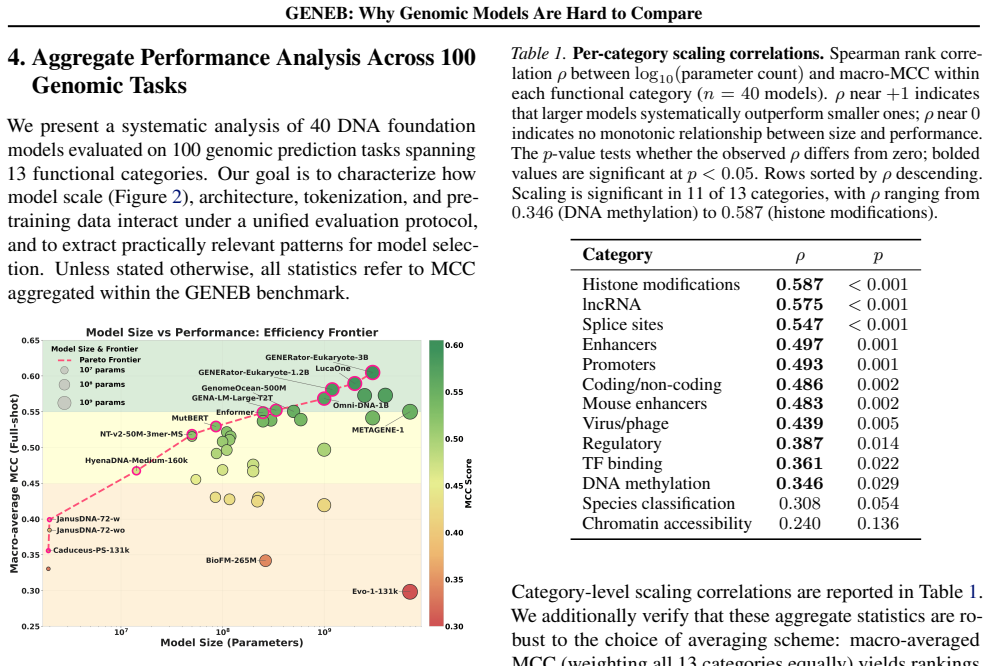
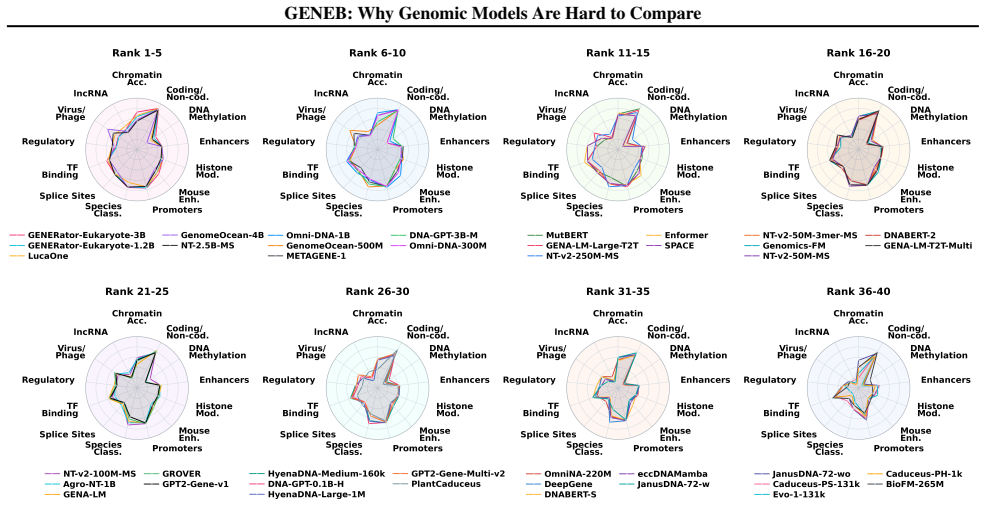
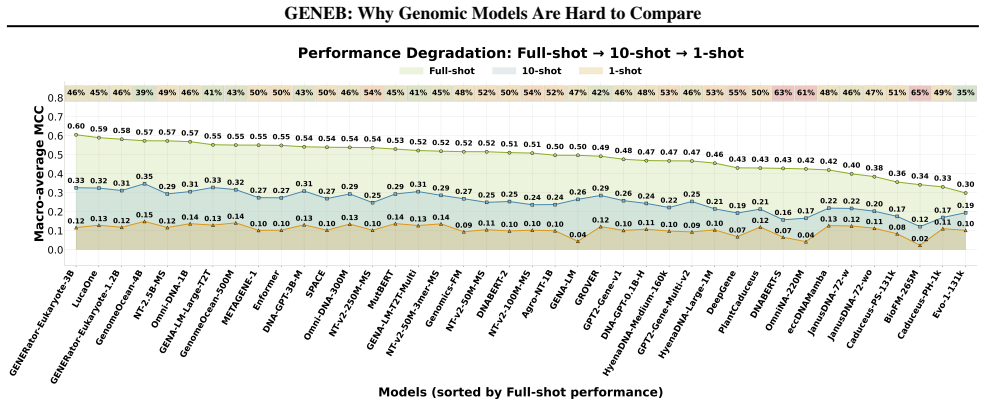
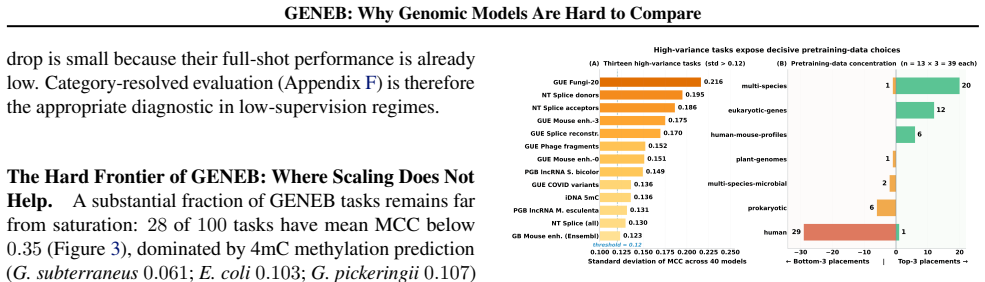
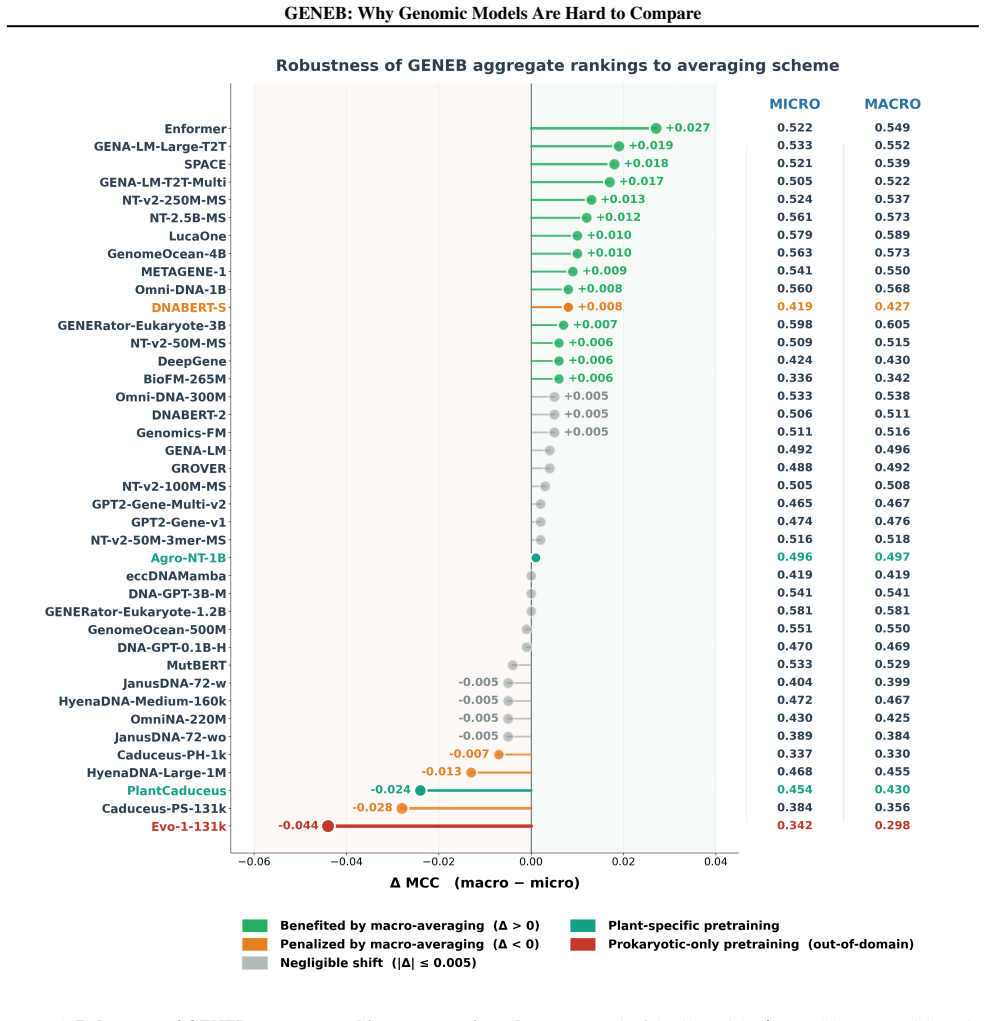
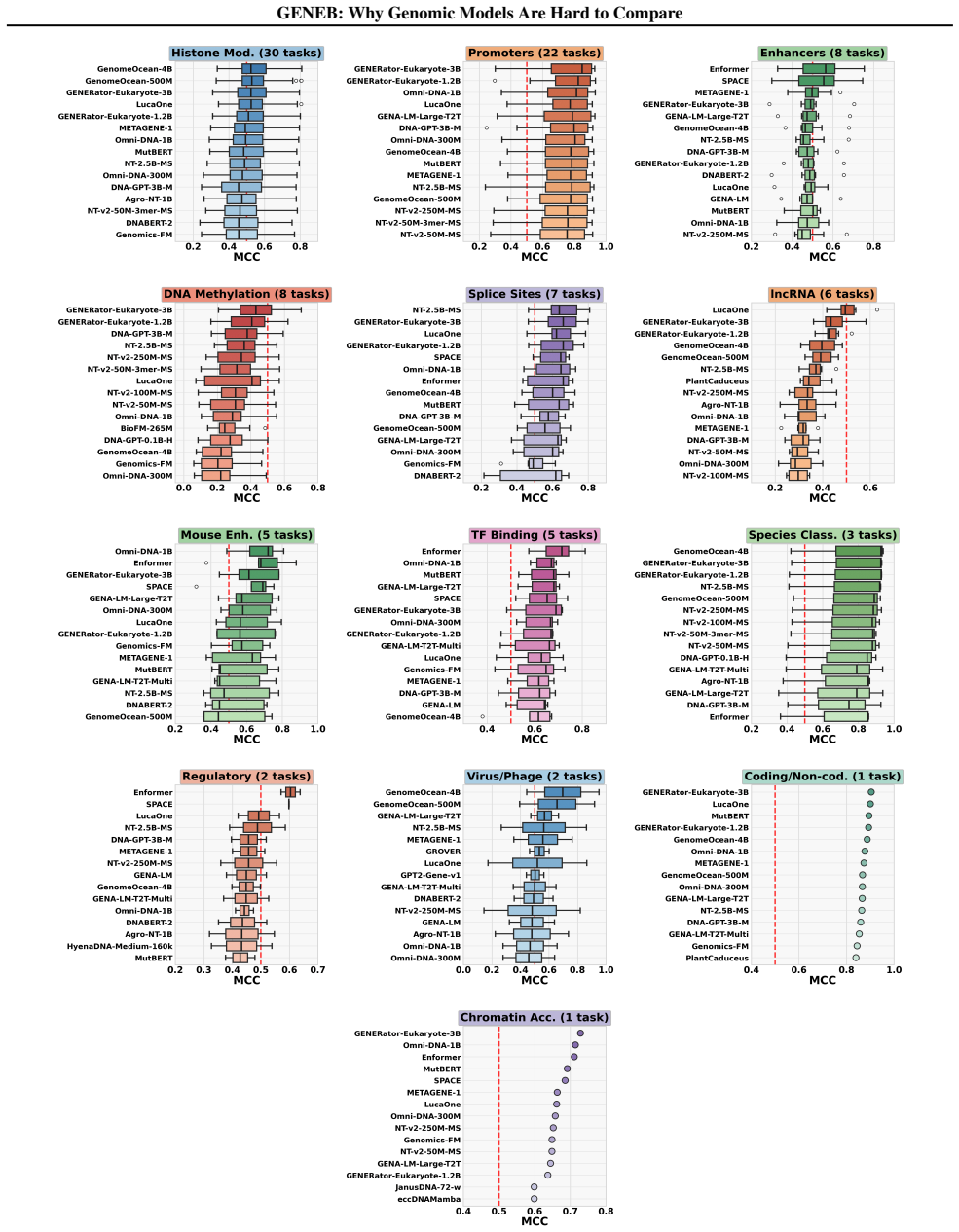
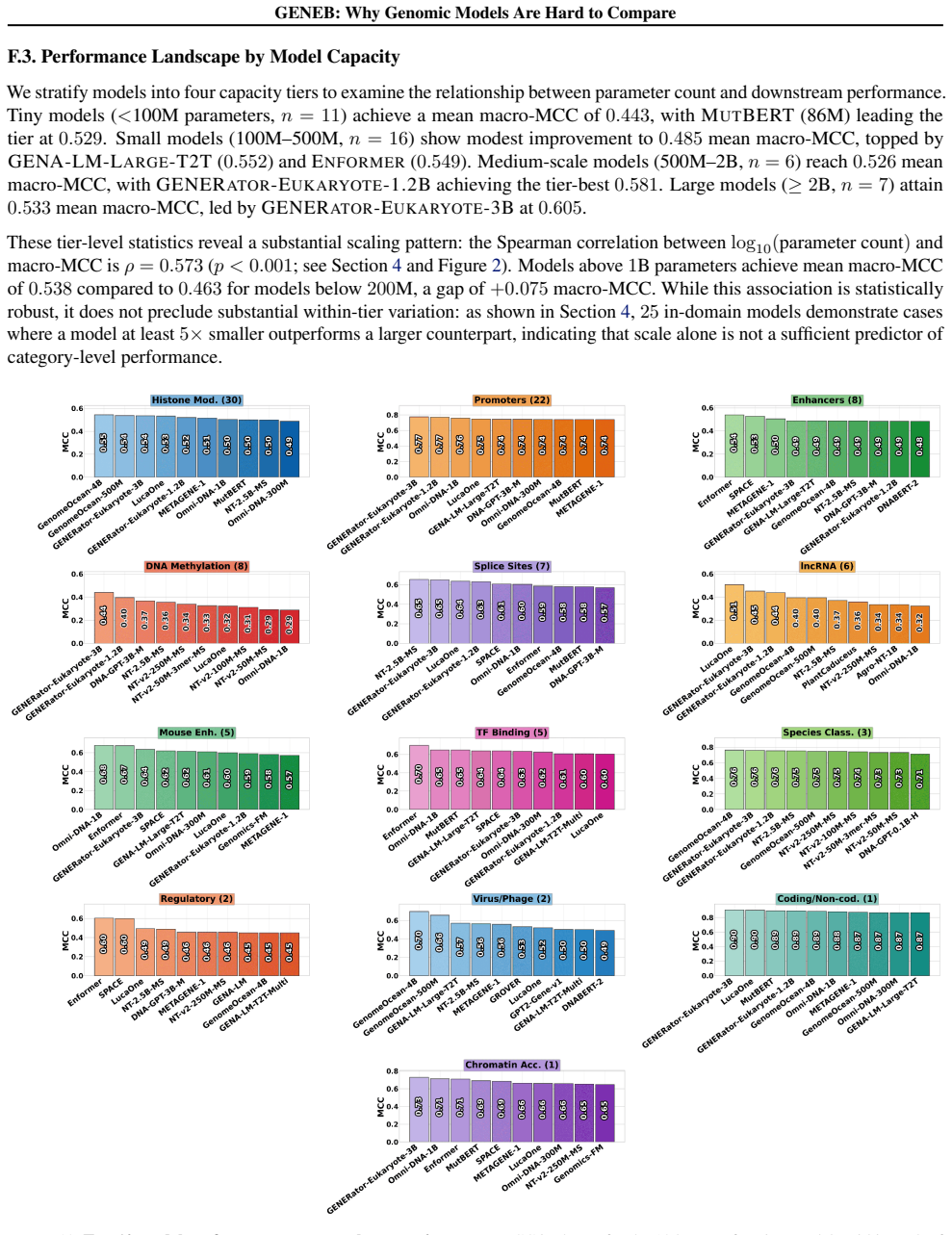
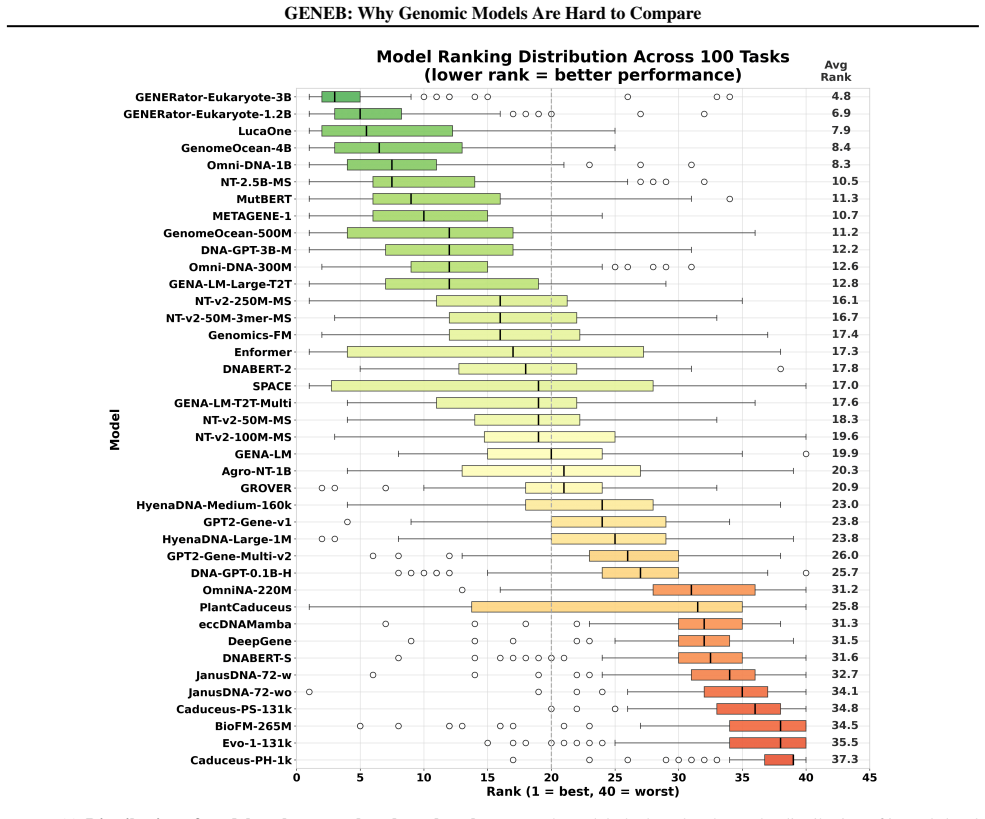
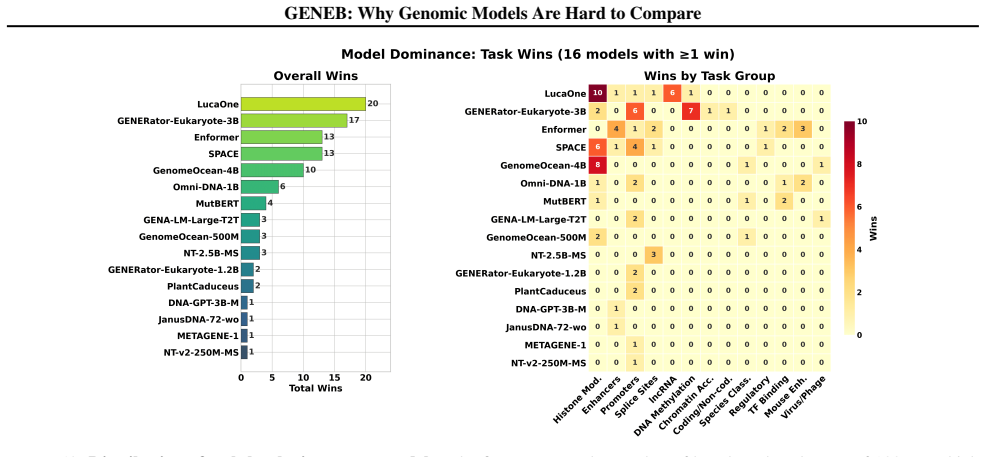
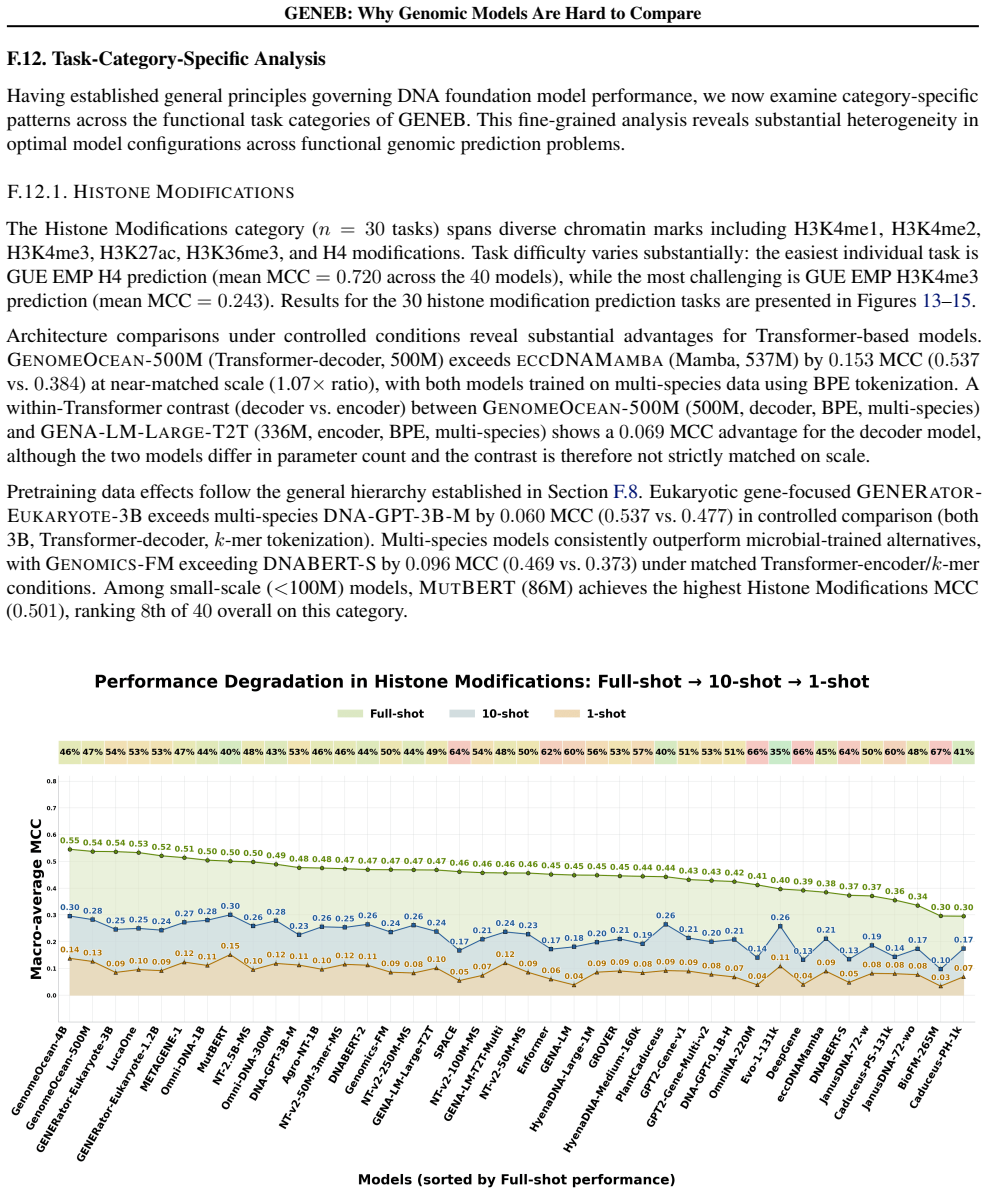
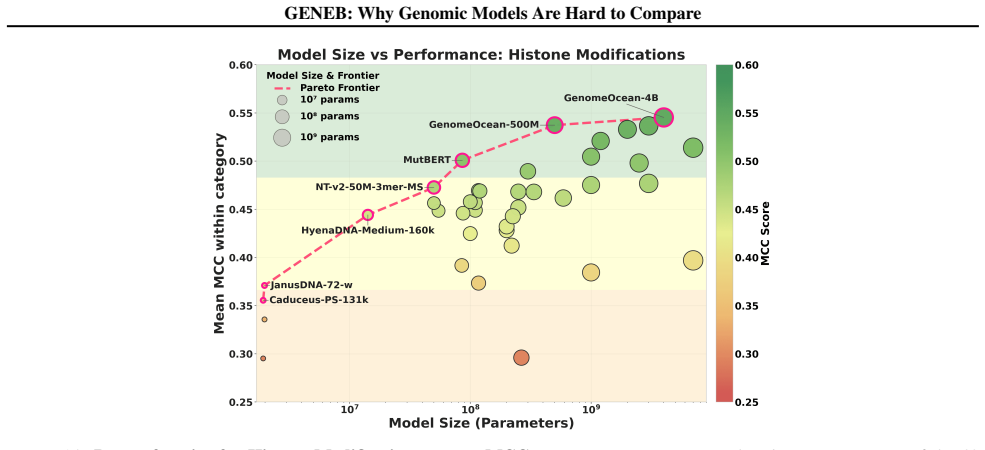
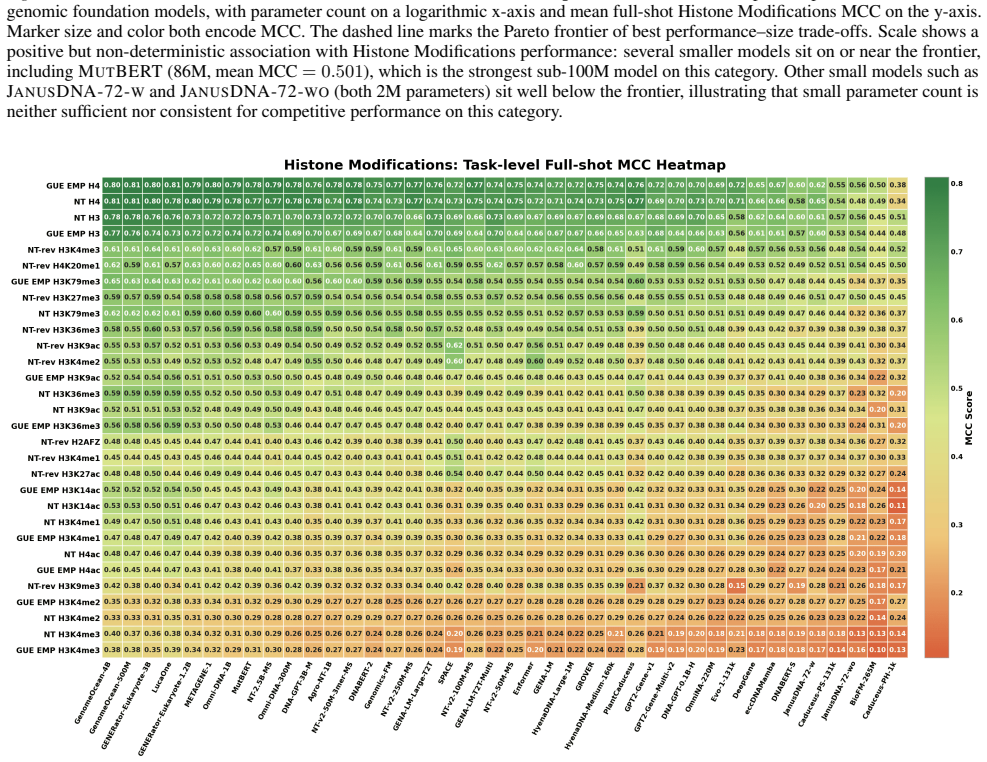
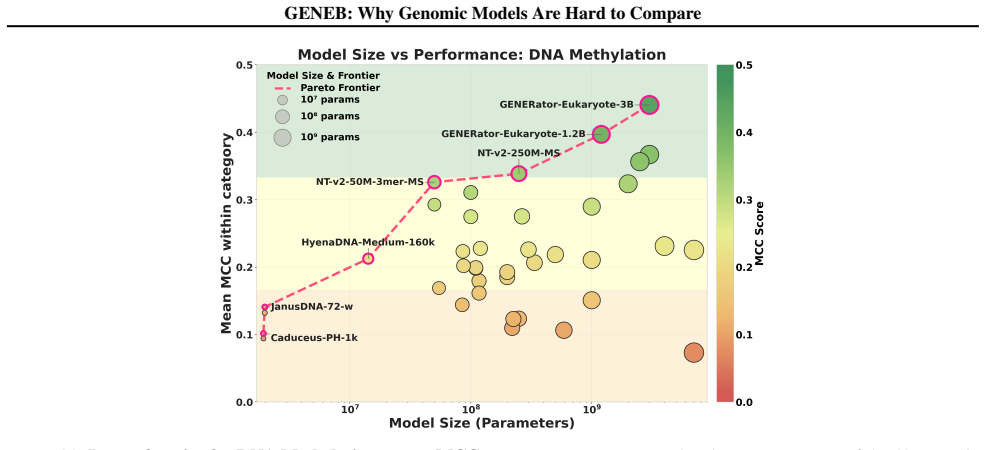
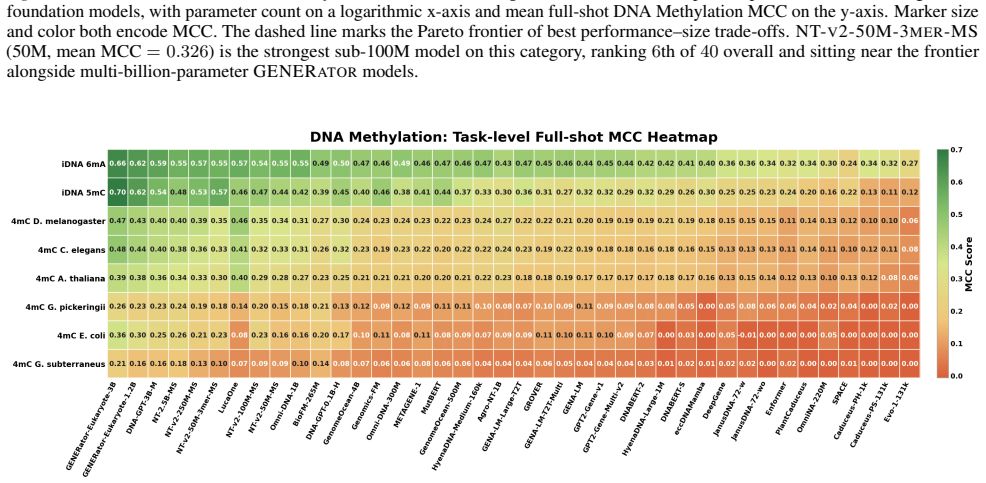
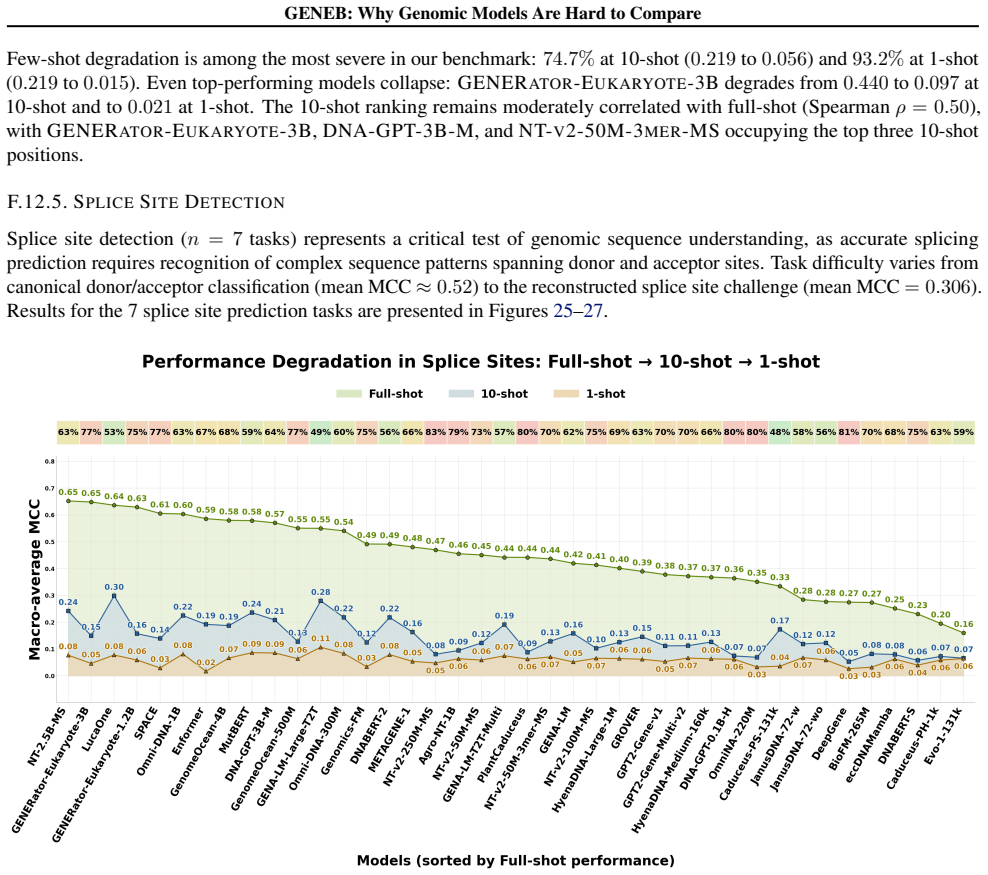
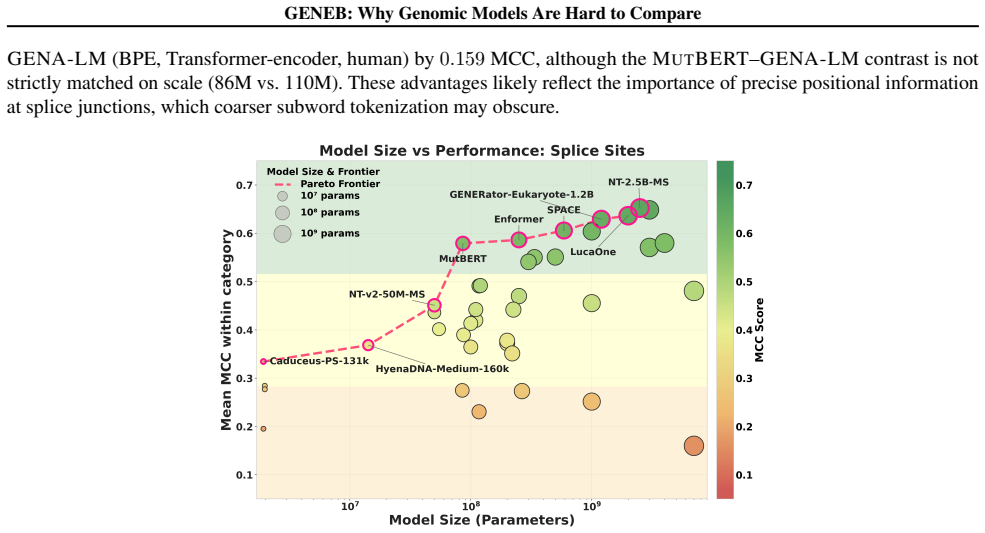
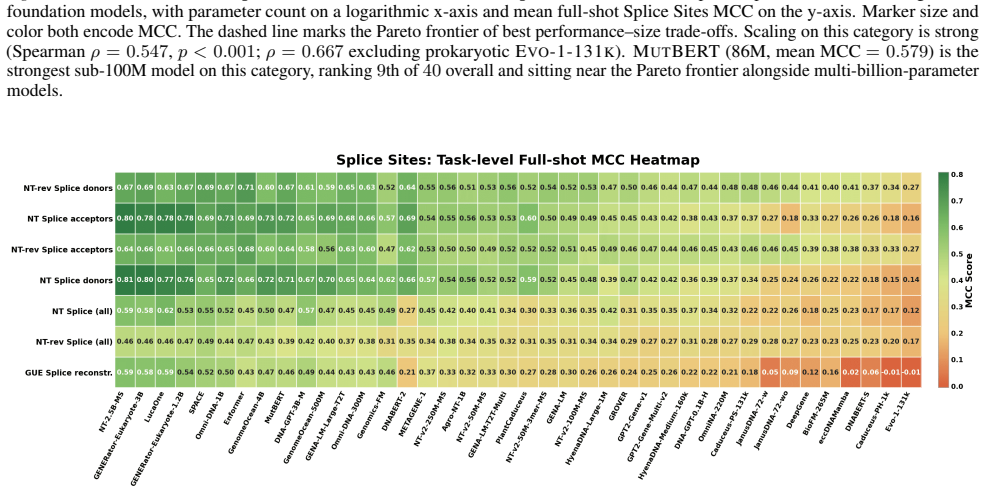
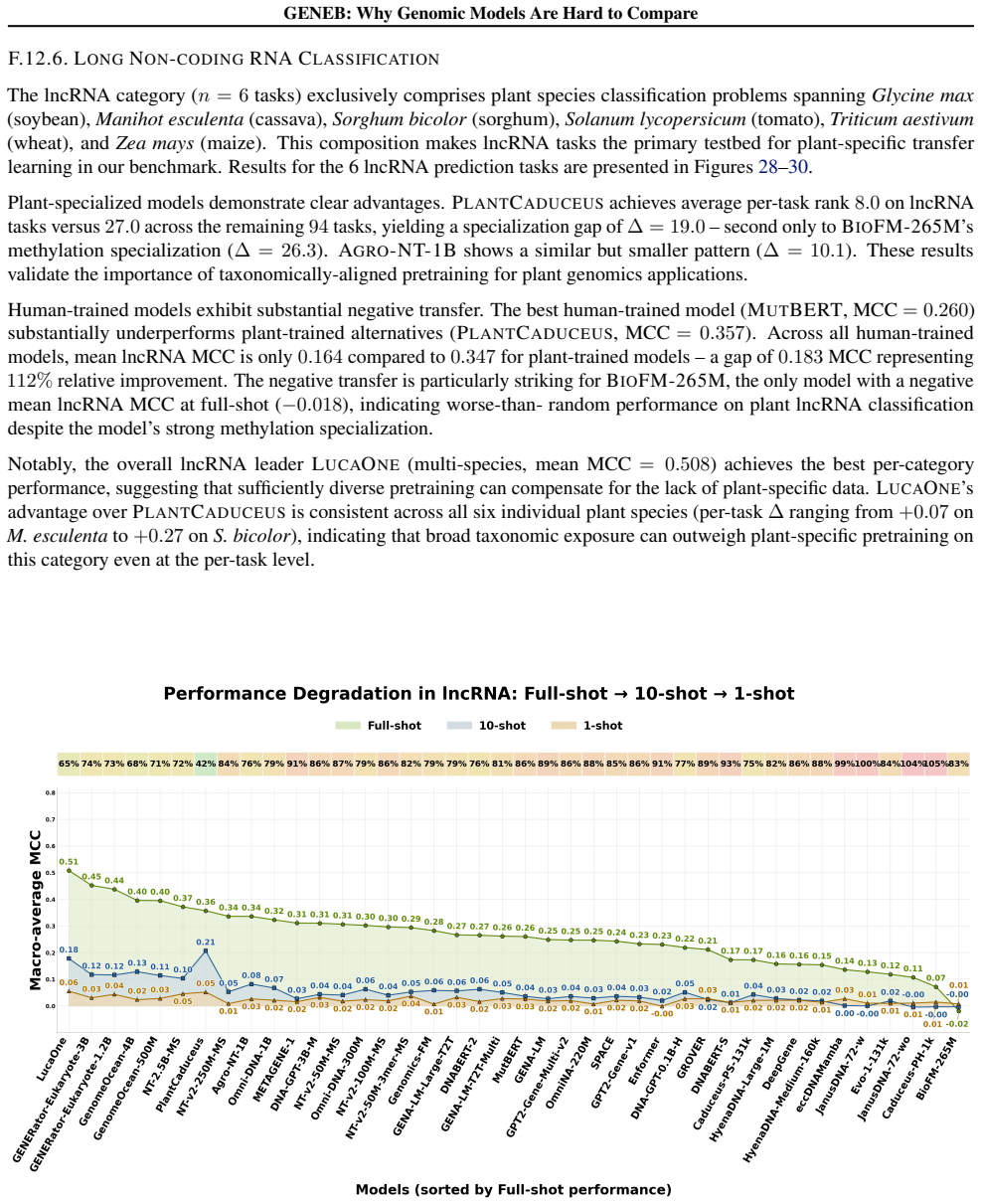
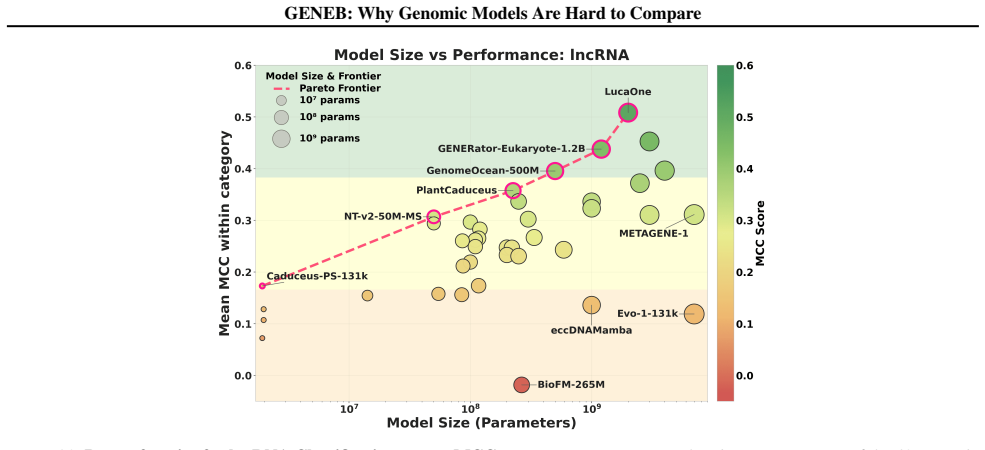
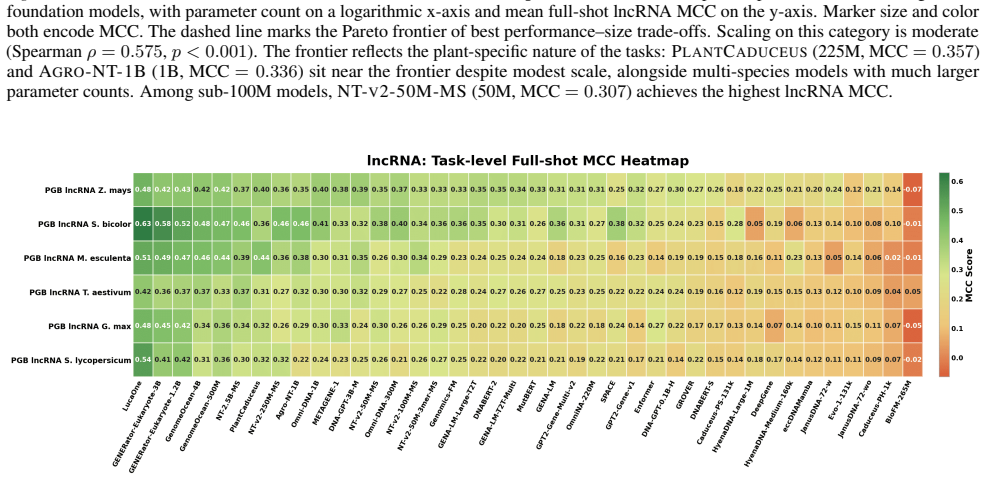
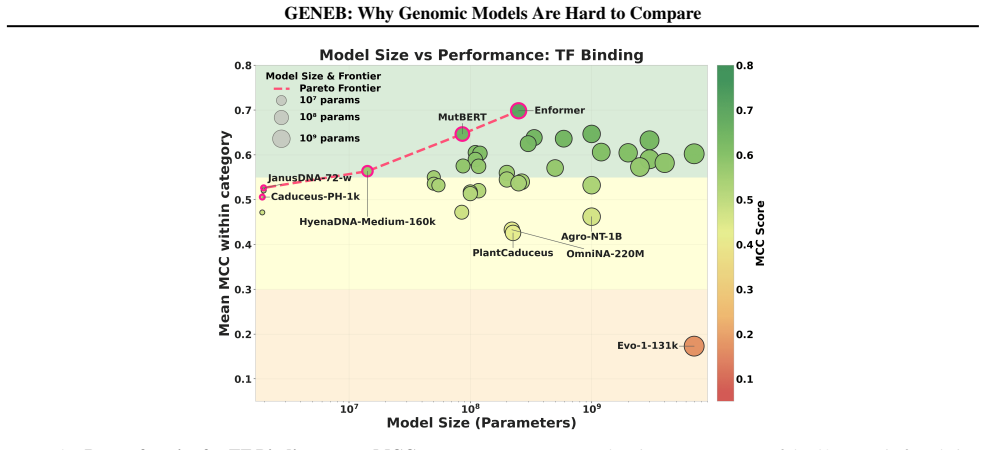
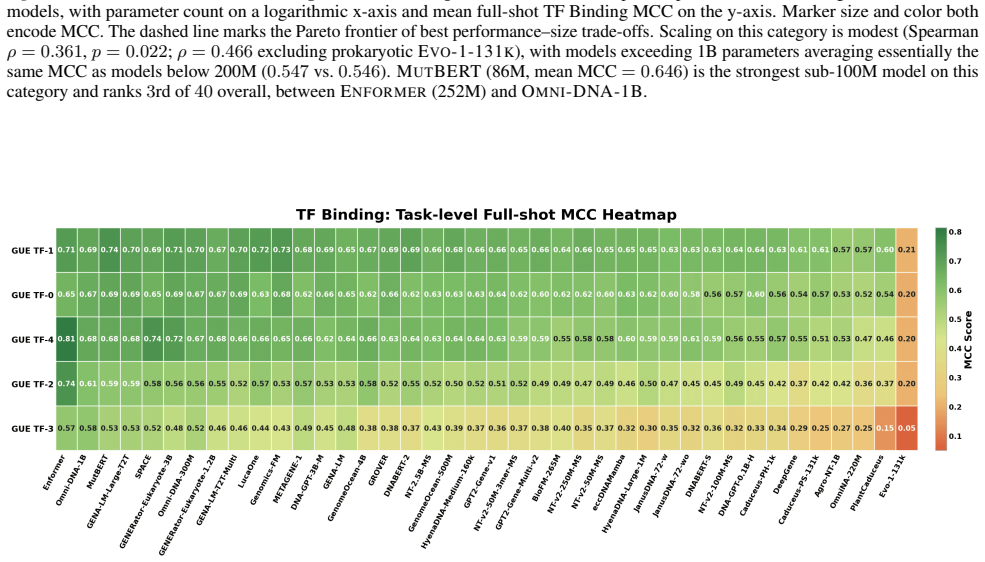
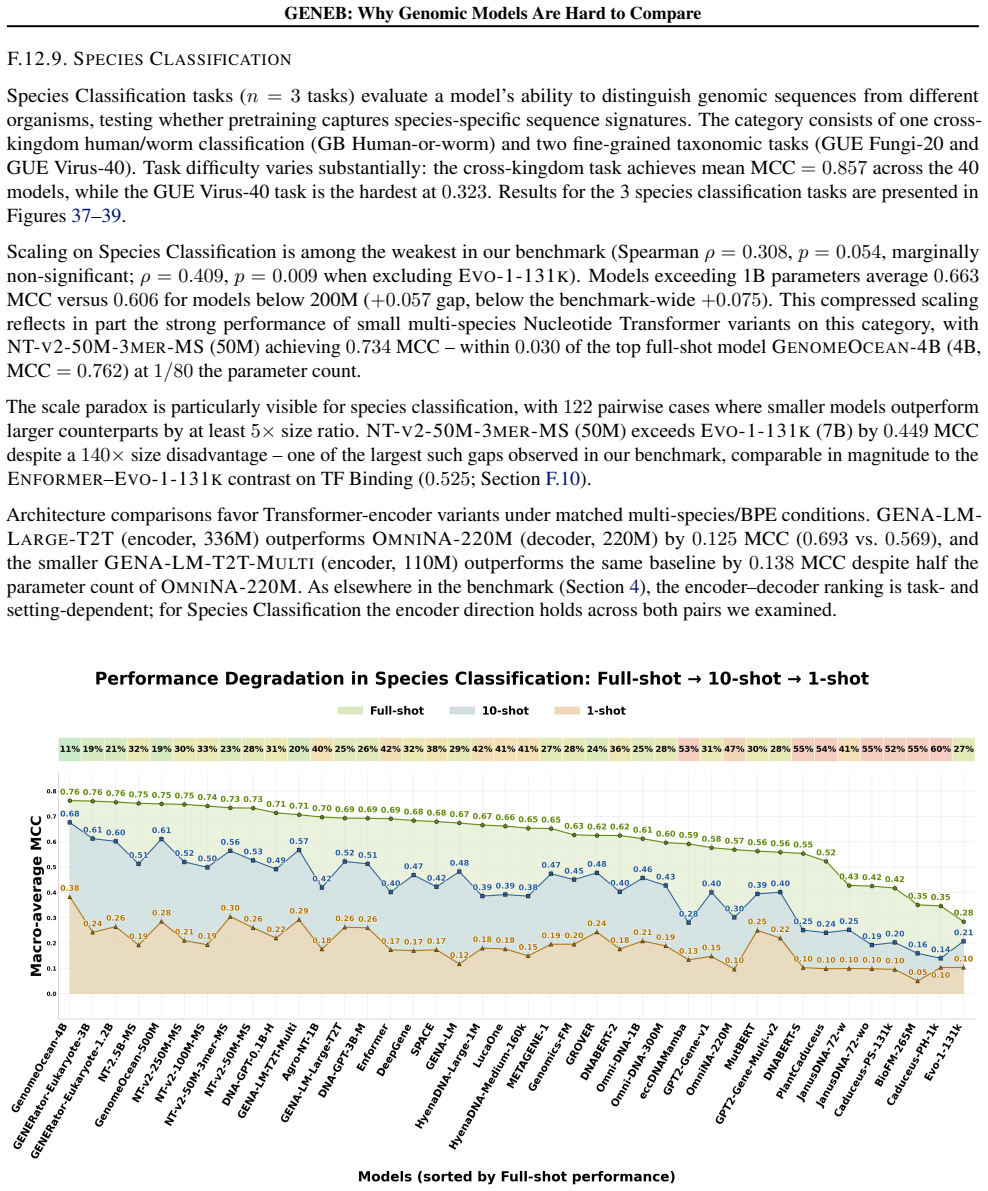
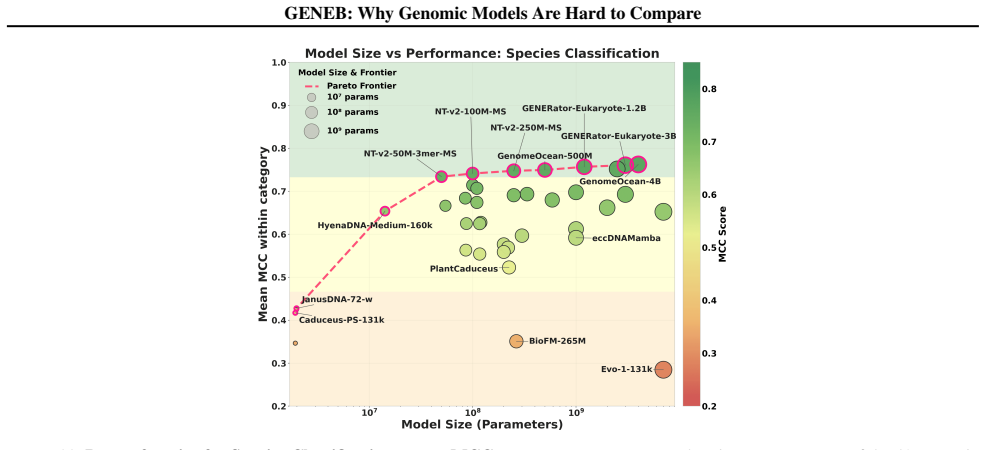
GENEB applies a unified probing-based protocol to frozen representations from 40 genomic foundation models across 100 tasks in 13 functional categories and finds that aggregate leaderboards are unstable, with rankings varying sharply across categories, scale providing only modest and inconsistent gains, and architectural and pretraining alignment frequently outweighing parameter count.
What carries the argument
GENEB, the diagnostic benchmark that runs a single probing protocol on frozen representations across 100 genomic tasks in 13 categories to expose task-level trade-offs.
Load-bearing premise
That one probing protocol on frozen representations across many tasks produces fair comparisons of true model capabilities without task-specific fine-tuning.
What would settle it
Re-running the 40 models with task-specific fine-tuning and finding that scale then dominates or rankings stabilize would undermine the claim that architecture outweighs scale under the frozen protocol.
Figures


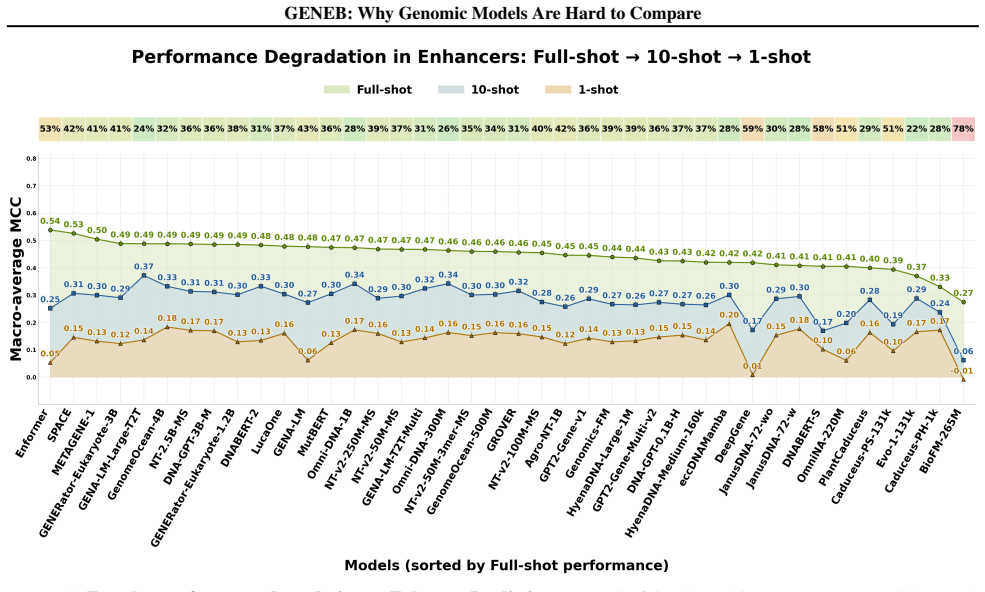
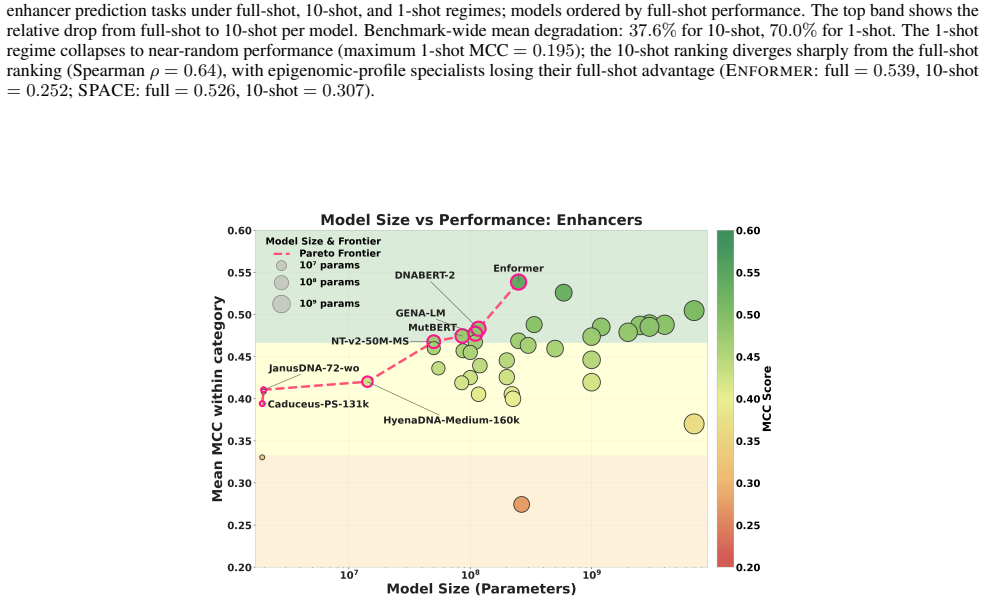
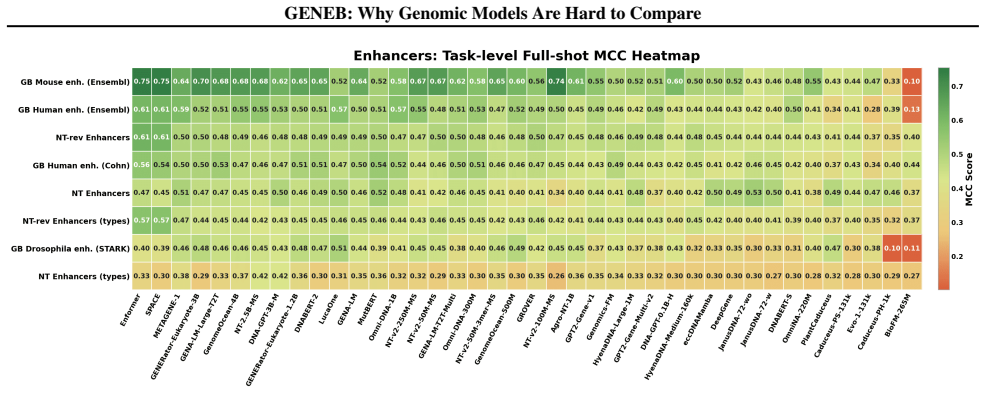
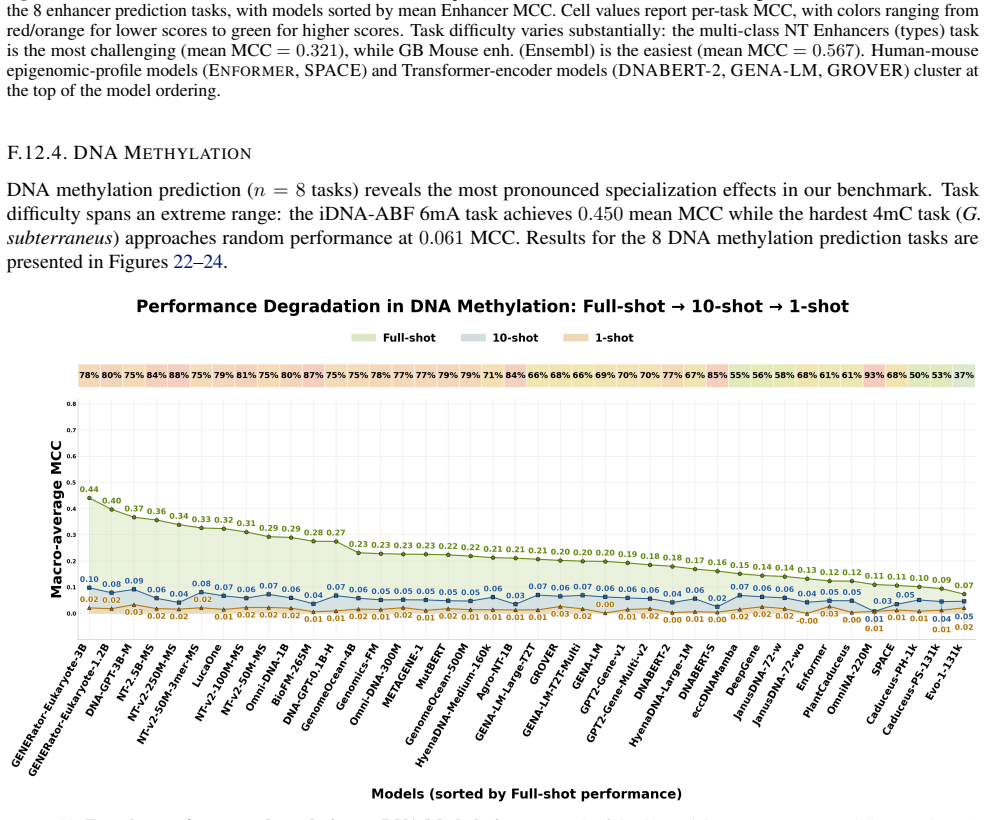
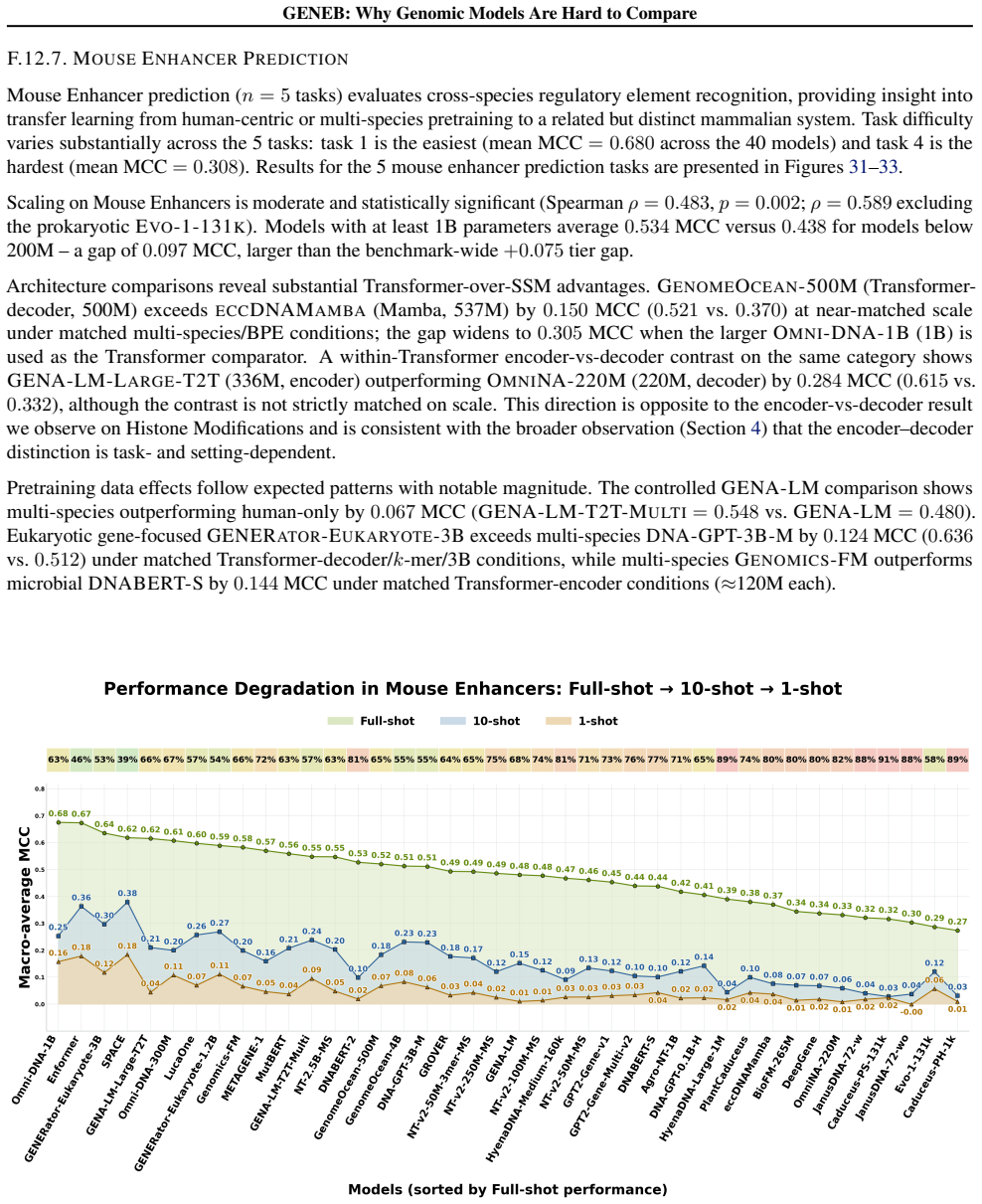
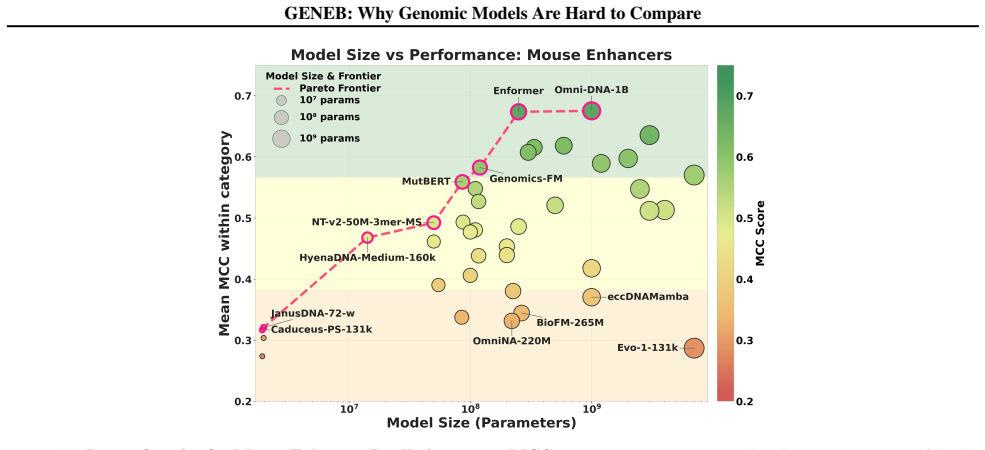
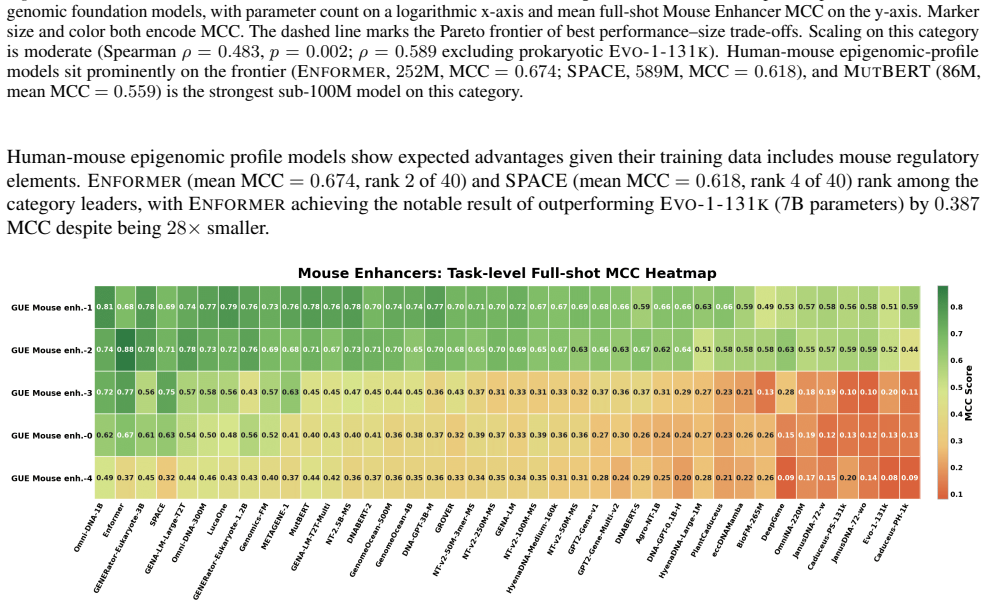
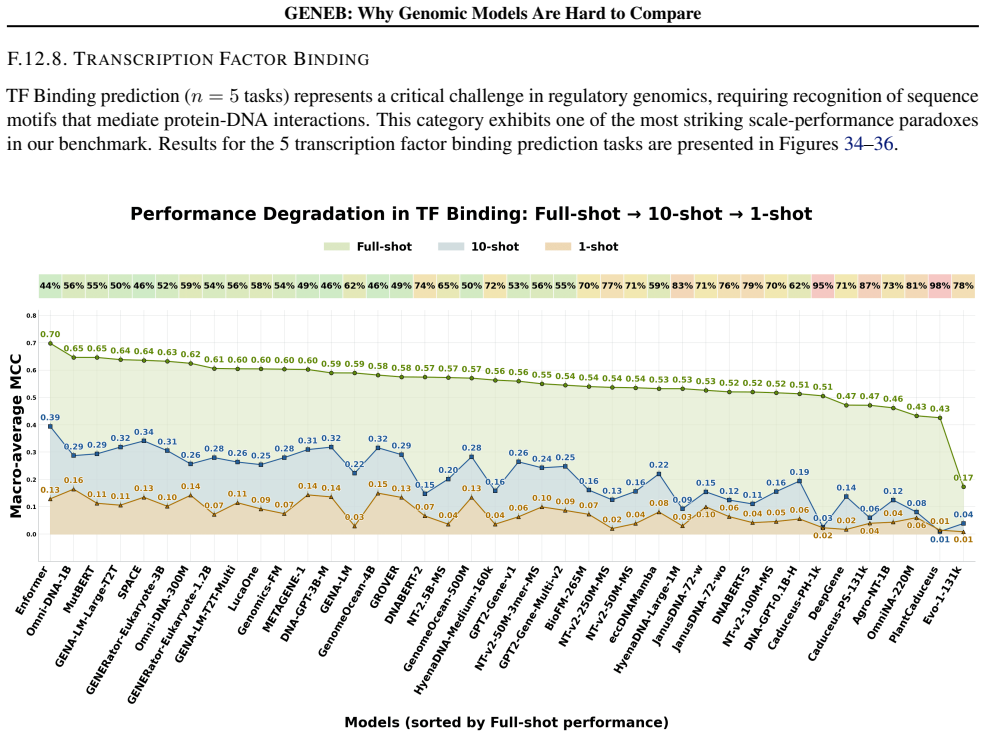
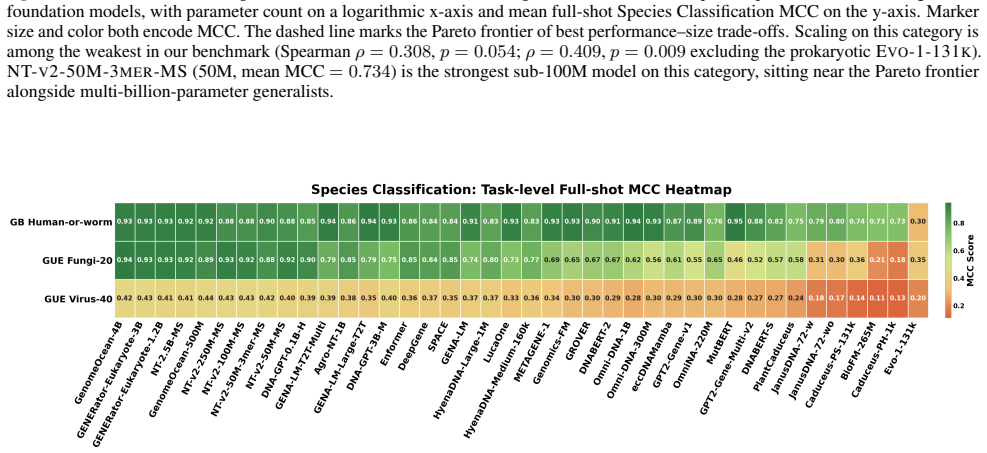
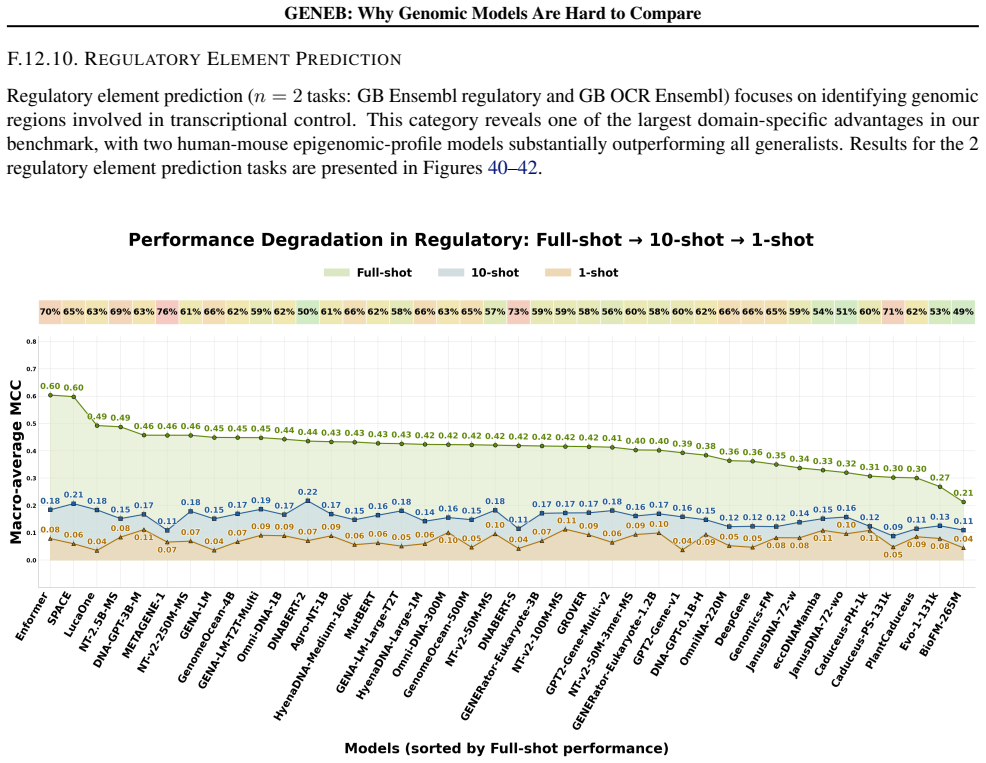

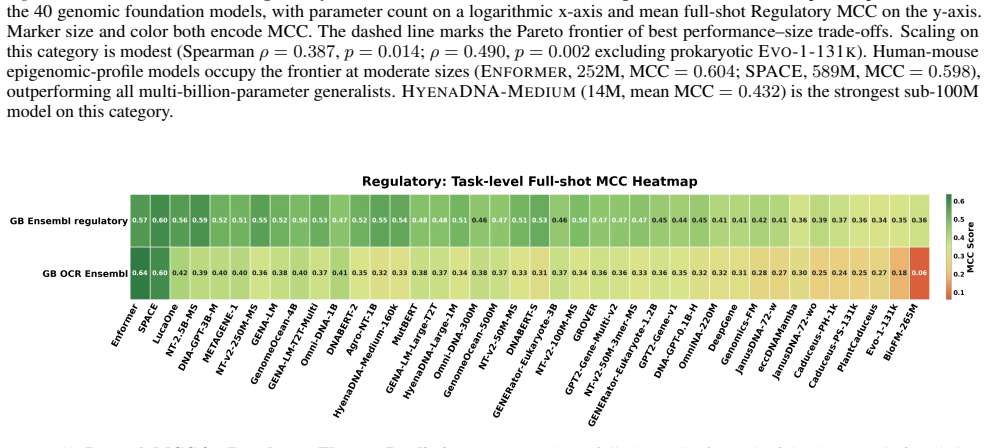
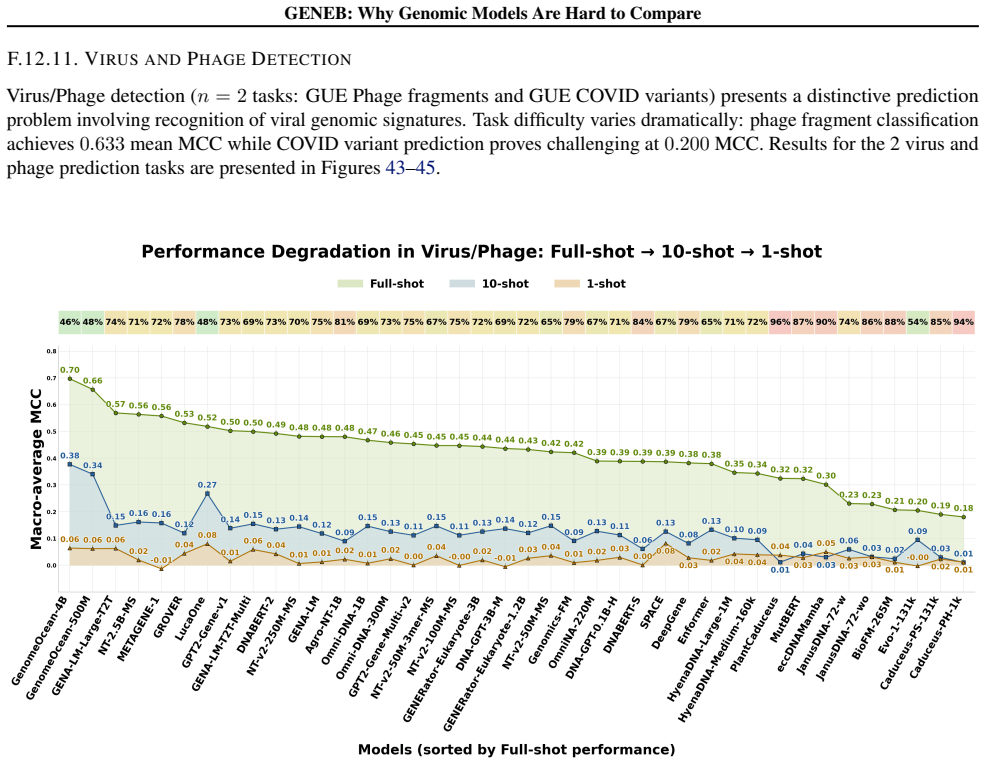

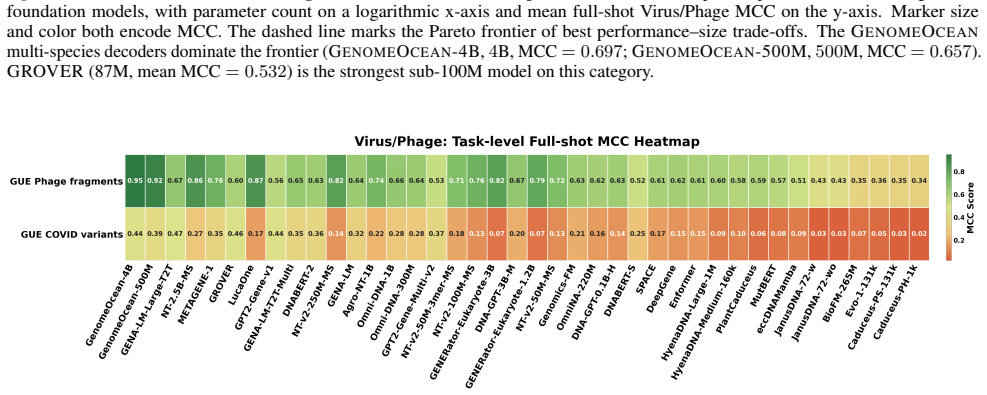
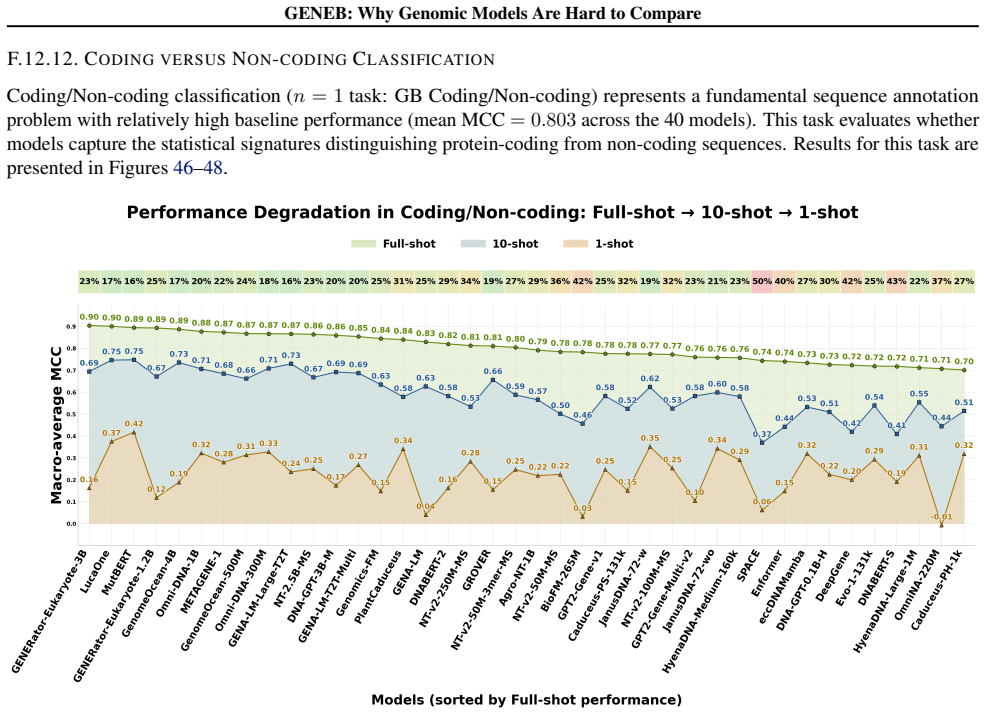
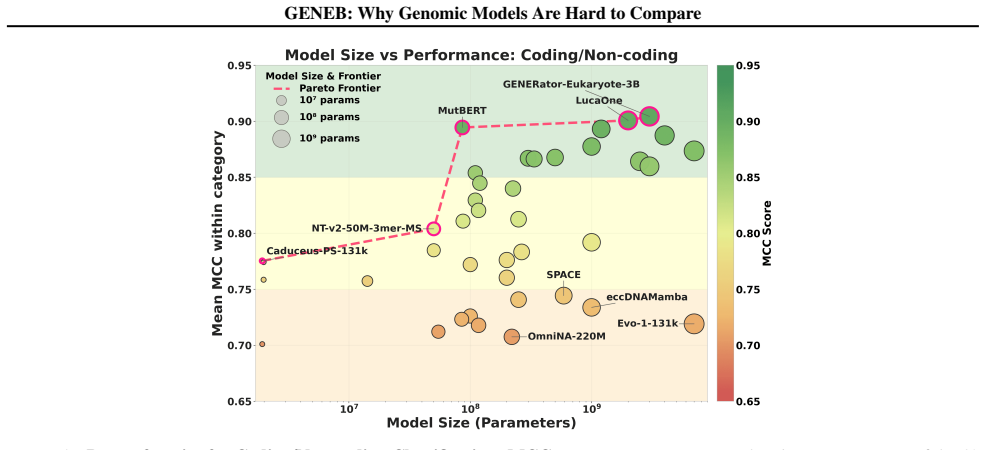
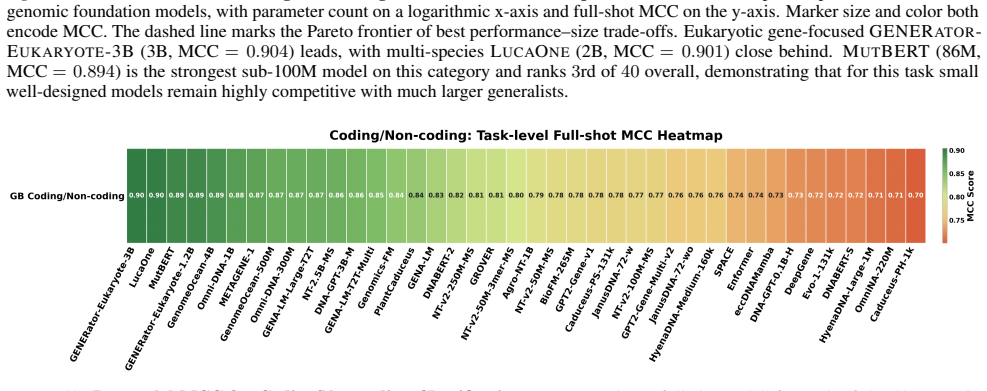
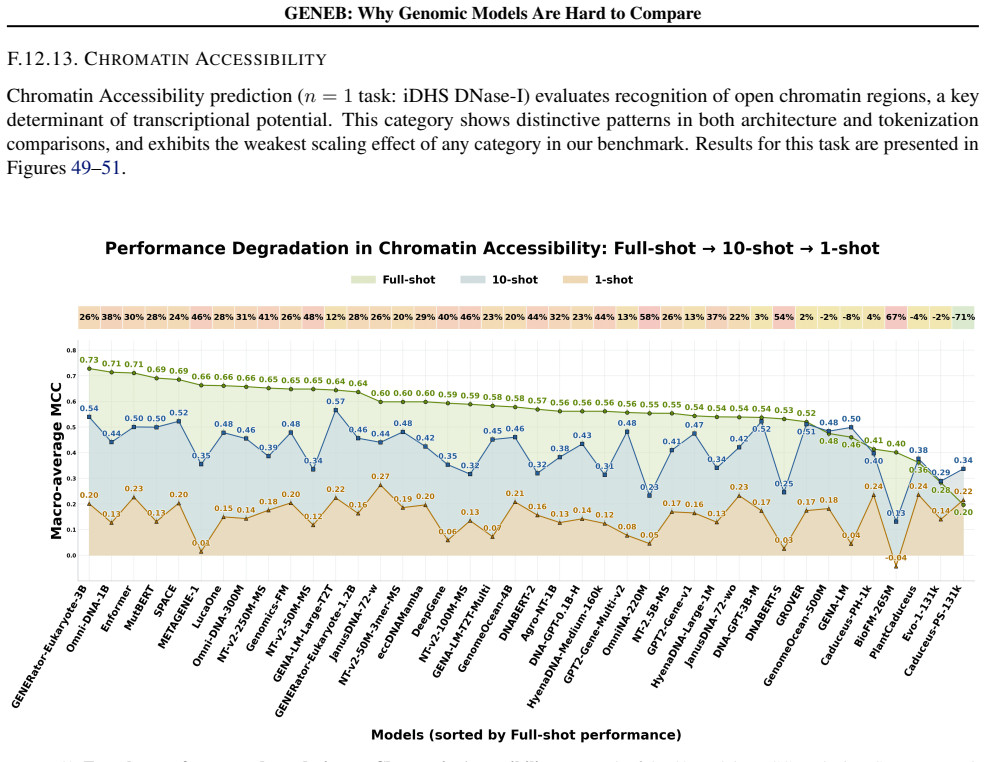
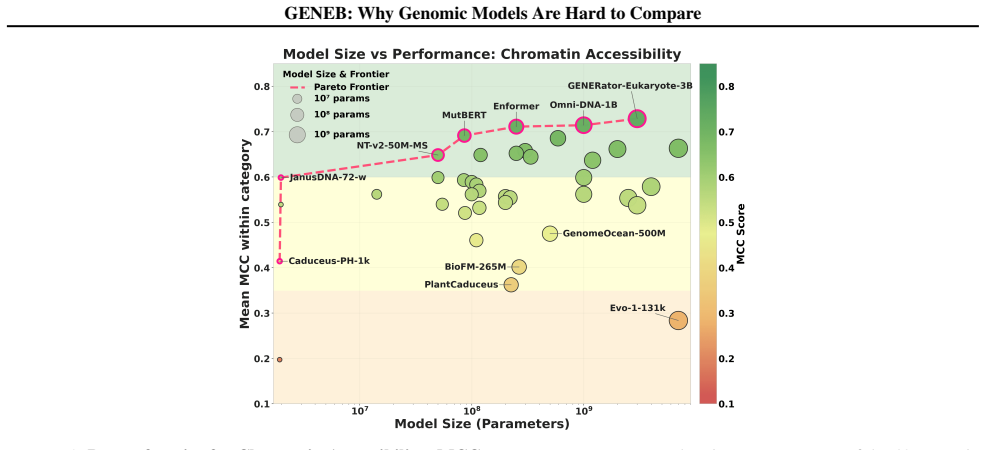
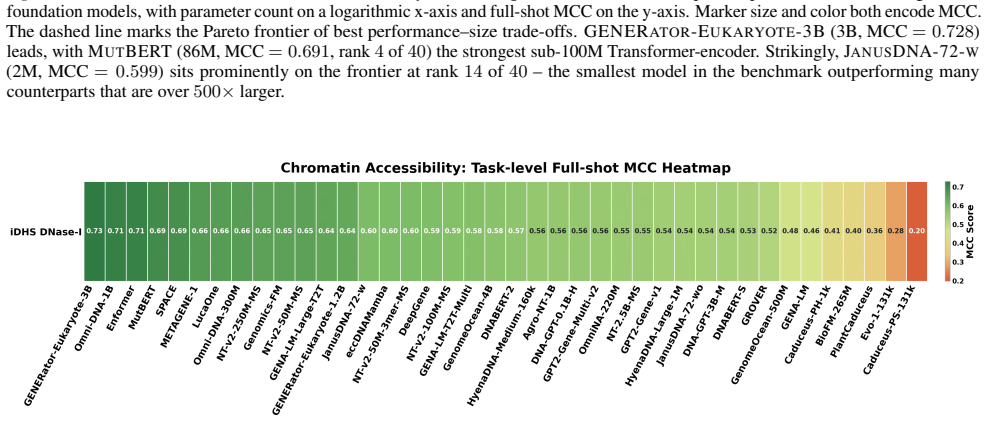
read the original abstract
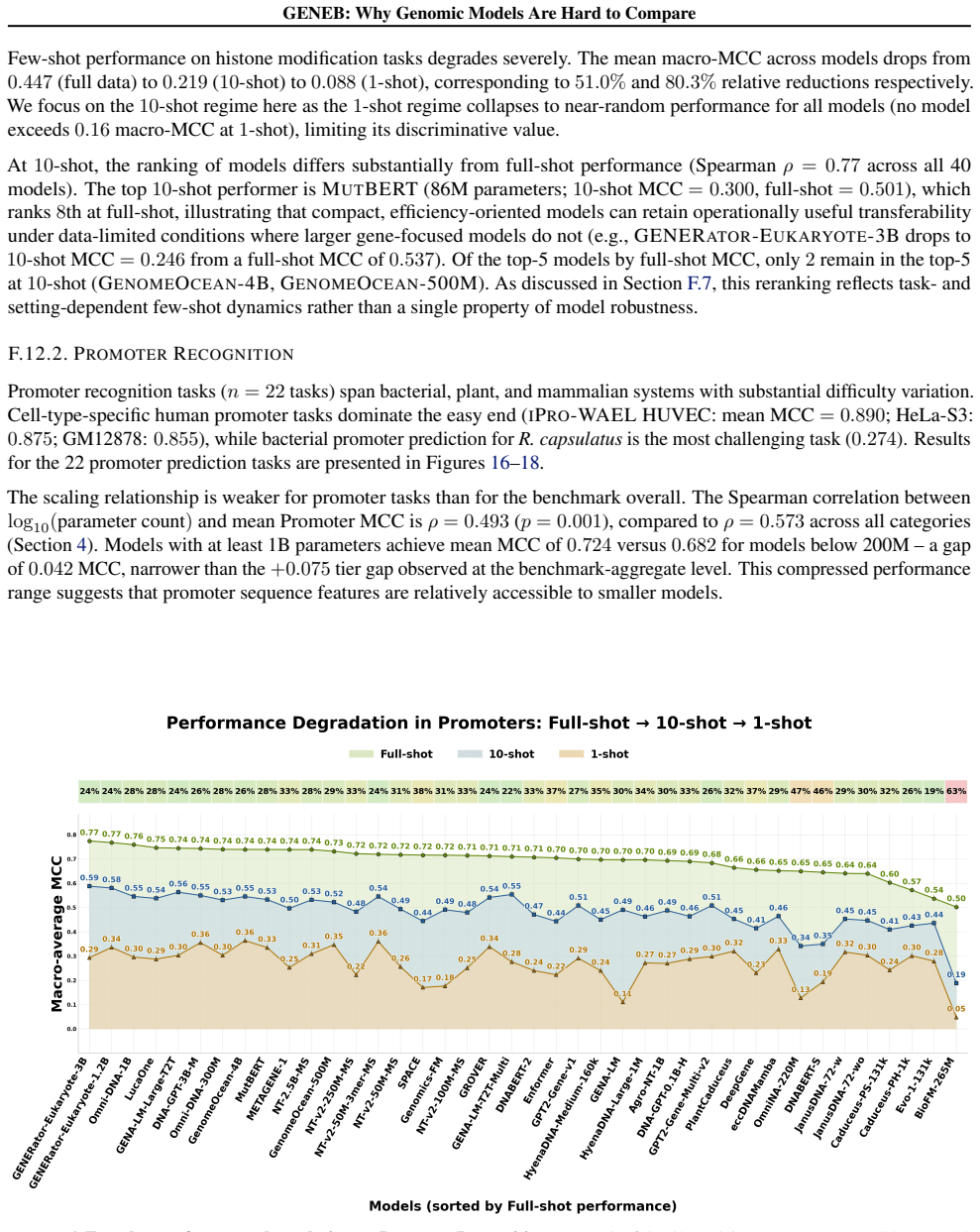
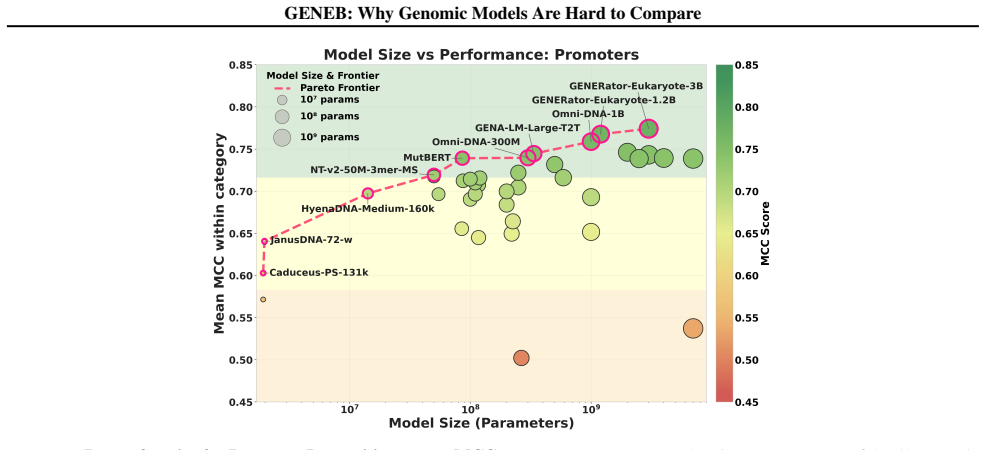
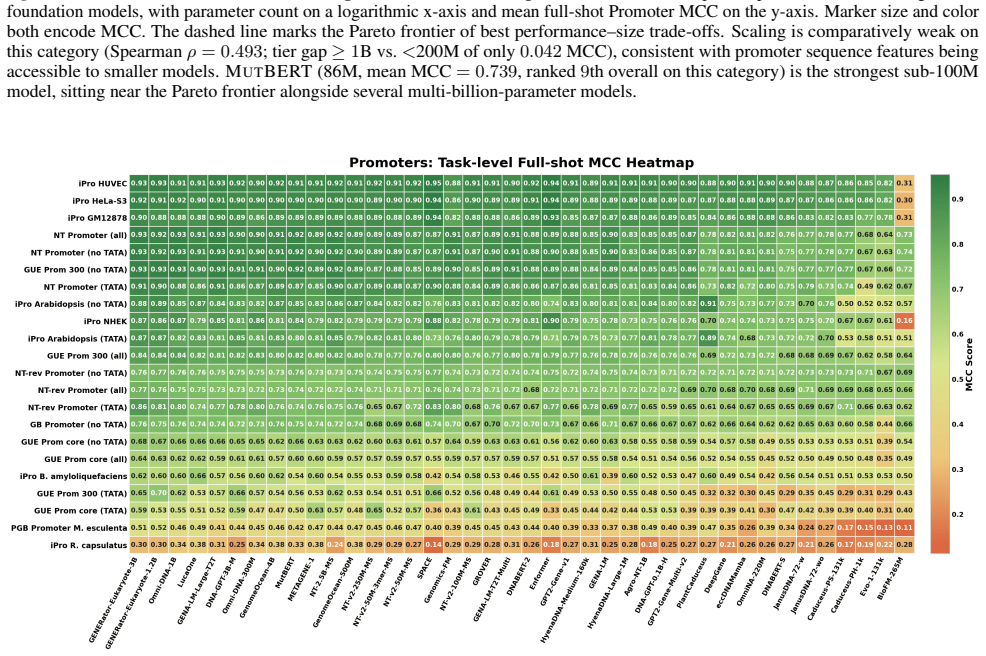
Progress in genomic foundation models is difficult to assess due to fragmented benchmarks, incompatible evaluation protocols, and task-specific reporting. As a result, claims of superiority or generality across models are often not directly comparable. We introduce GENEB, a large-scale diagnostic benchmark that evaluates frozen representations from 40 genomic foundation models across 100 tasks spanning 13 functional categories under a unified probing-based protocol, including few-shot regimes. GENEB enables controlled comparison across model scale, architecture, tokenization, and pretraining data while explicitly exposing task-level trade-offs. Our analysis shows that aggregate leaderboards are unstable: model rankings vary sharply across task categories, scale provides only modest and inconsistent gains, and architectural and pretraining alignment frequently outweigh parameter count. These results highlight limitations of current evaluation practices and position GENEB as a reference framework for principled comparison and category-aware model selection in genomic machine learning.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces GENEB, a diagnostic benchmark that applies a unified probing protocol to frozen representations from 40 genomic foundation models across 100 tasks in 13 functional categories (including few-shot settings). It claims that aggregate leaderboards are unstable because model rankings vary sharply across task categories, that scale yields only modest and inconsistent gains, and that architectural and pretraining alignment frequently outweigh parameter count.
Significance. If the empirical findings are robust, the work provides concrete evidence of the limitations of current fragmented evaluation practices in genomic ML and supplies a controlled reference framework that could support category-aware model selection and more reliable comparisons.
major comments (2)
- [Evaluation protocol (unified probing description)] The central claims (leaderboard instability, modest scale effects, and architecture/pretraining dominance) rest entirely on orderings produced by one fixed probing protocol applied to frozen representations. No ablation or sensitivity analysis is reported for alternative probes (linear vs. MLP, regularization strength, pooling method), so it remains possible that the observed instabilities and trade-offs are artifacts of that specific protocol rather than intrinsic model properties.
- [Results and analysis sections] The manuscript does not report variance estimates, statistical significance tests, or confidence intervals on the ranking variations across the 13 categories or on the scale-gain inconsistencies; without these, it is difficult to determine whether the reported instabilities are load-bearing or could arise from task sampling or probe variance.
minor comments (2)
- Provide an explicit list or table of all 40 models, their scales, architectures, tokenizers, and pretraining corpora to enable direct replication of the controlled comparisons.
- Clarify the precise definition and implementation of the 'few-shot regimes' (number of shots, sampling strategy, and how they differ from the main probing setup).
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive comments. The feedback highlights important aspects of robustness that we will address in revision. We respond point-by-point to the major comments below.
read point-by-point responses
-
Referee: The central claims (leaderboard instability, modest scale effects, and architecture/pretraining dominance) rest entirely on orderings produced by one fixed probing protocol applied to frozen representations. No ablation or sensitivity analysis is reported for alternative probes (linear vs. MLP, regularization strength, pooling method), so it remains possible that the observed instabilities and trade-offs are artifacts of that specific protocol rather than intrinsic model properties.
Authors: The unified fixed probing protocol was deliberately chosen to isolate model-intrinsic differences (architecture, scale, pretraining alignment) by removing confounding variation from the evaluation method itself; varying the probe would undermine the controlled comparison that GENEB is designed to provide. We acknowledge that probe sensitivity is a valid concern for generalizability. In the revised manuscript we will add a targeted sensitivity analysis on a representative subset of tasks and models, comparing linear vs. MLP probes and alternative pooling/regularization choices, to quantify how much the reported instabilities persist under protocol variation. revision: yes
-
Referee: The manuscript does not report variance estimates, statistical significance tests, or confidence intervals on the ranking variations across the 13 categories or on the scale-gain inconsistencies; without these, it is difficult to determine whether the reported instabilities are load-bearing or could arise from task sampling or probe variance.
Authors: We agree that statistical quantification of the ranking instability and scale effects would strengthen the claims. The revised version will include bootstrap-derived confidence intervals on per-category rankings and on the scale-gain deltas, together with permutation tests assessing whether observed rank changes across categories exceed what would be expected from task sampling variance alone. revision: yes
Circularity Check
No circularity; empirical observations only
full rationale
The paper introduces GENEB as an empirical benchmark and reports direct observations from running a fixed probing protocol on 40 models across 100 tasks. No equations, derivations, fitted parameters, or self-citations appear as load-bearing steps in any claimed result. All central claims (unstable rankings, modest scale gains, architecture outweighing parameter count) are presented as outcomes of the benchmark execution itself, with no reduction to prior inputs by construction. This is the standard case of a self-contained empirical study.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Effective gene expression prediction from sequence by integrating long-range interactions , volume =
Avsec, Ziga and Agarwal, Vikram and Visentin, Daniel and Ledsam, Joseph and Grabska-Barwinska, Agnieszka and Taylor, Kyle and Assael, Yannis and Jumper, John and Kohli, Pushmeet and Kelley, David , year =. Effective gene expression prediction from sequence by integrating long-range interactions , volume =. Nature Methods , doi =
-
[2]
2024 , eprint=
DNABERT-2: Efficient Foundation Model and Benchmark For Multi-Species Genome , author=. 2024 , eprint=
2024
-
[3]
2023 , eprint=
MTEB: Massive Text Embedding Benchmark , author=. 2023 , eprint=
2023
-
[4]
2024 , eprint=
DNABERT-S: Pioneering Species Differentiation with Species-Aware DNA Embeddings , author=. 2024 , eprint=
2024
-
[5]
2025 , eprint=
GENERator: A Long-Context Generative Genomic Foundation Model , author=. 2025 , eprint=
2025
-
[6]
DNA language model GROVER learns sequence context in the human genome , volume =
Sanabria, Melissa and Hirsch, Jonas and Joubert, Pierre and Poetsch, Anna , year =. DNA language model GROVER learns sequence context in the human genome , volume =. Nature Machine Intelligence , doi =
-
[7]
2025 , doi =
Zhou, Zhihan and Riley, Robert and Kautsar, Satria and Wu, Weimin and Egan, Rob and Hofmeyr, Steven and Goldhaber-Gordon, Shira and Yu, Mutian and Ho, Harrison and Liu, Fengchen and Chen, Feng and Morgan-Kiss, Rachael and Shi, Lizhen and Liu, Han and Wang, Zhong , title =. 2025 , doi =. https://www.biorxiv.org/content/early/2025/02/05/2025.01.30.635558.fu...
2025
-
[8]
2025 , eprint=
JanusDNA: A Powerful Bi-directional Hybrid DNA Foundation Model , author=. 2025 , eprint=
2025
-
[9]
2025 , eprint=
METAGENE-1: Metagenomic Foundation Model for Pandemic Monitoring , author=. 2025 , eprint=
2025
-
[10]
2025 , eprint=
Omni-DNA: A Unified Genomic Foundation Model for Cross-Modal and Multi-Task Learning , author=. 2025 , eprint=
2025
-
[11]
bioRxiv , pages=
A Foundational Large Language Model for Edible Plant Genomes , author=. bioRxiv , pages=. 2023 , publisher=
2023
-
[12]
2024 , eprint=
Caduceus: Bi-Directional Equivariant Long-Range DNA Sequence Modeling , author=. 2024 , eprint=
2024
-
[13]
2023 , eprint=
DNAGPT: A Generalized Pre-trained Tool for Versatile DNA Sequence Analysis Tasks , author=. 2023 , eprint=
2023
-
[14]
2025 , eprint=
eccDNAMamba: A Pre-Trained Model for Ultra-Long eccDNA Sequence Analysis , author=. 2025 , eprint=
2025
-
[15]
Durrant and Brian Kang and Dhruva Katrekar and David B
Eric Nguyen and Michael Poli and Matthew G. Durrant and Brian Kang and Dhruva Katrekar and David B. Li and Liam J. Bartie and Armin W. Thomas and Samuel H. King and Garyk Brixi and Jeremy Sullivan and Madelena Y. Ng and Ashley Lewis and Aaron Lou and Stefano Ermon and Stephen A. Baccus and Tina Hernandez-Boussard and Christopher Ré and Patrick D. Hsu and ...
2024
-
[16]
2023 , doi =
Veniamin Fishman and Yuri Kuratov and Maxim Petrov and Aleksei Shmelev and Denis Shepelin and Nikolay Chekanov and Olga Kardymon and Mikhail Burtsev , title =. 2023 , doi =. https://www.biorxiv.org/content/early/2023/06/13/2023.06.12.544594.full.pdf , journal =
2023
-
[17]
2023 , eprint=
HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution , author=. 2023 , eprint=
2023
-
[18]
bioRxiv , pages=
The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics , author=. bioRxiv , pages=. 2023 , publisher=
2023
-
[19]
2025 , eprint=
SPACE: Your Genomic Profile Predictor is a Powerful DNA Foundation Model , author=. 2025 , eprint=
2025
-
[20]
2024 , doi =
Shen, Xilin and Li, Xiangchun , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/01/15/2024.01.14.575543.full.pdf , journal =
2024
-
[21]
2024 , eprint=
BEND: Benchmarking DNA Language Models on biologically meaningful tasks , author=. 2024 , eprint=
2024
-
[22]
Miller and Armin Scheben and Michelle C
Jingjing Zhai and Aaron Gokaslan and Yair Schiff and Ana Berthel and Zong-Yan Liu and Wei-Yun Lai and Zachary R. Miller and Armin Scheben and Michelle C. Stitzer and M. Cinta Romay and Edward S. Buckler and Volodymyr Kuleshov , title =. Proceedings of the National Academy of Sciences , volume =. 2025 , doi =. https://www.pnas.org/doi/pdf/10.1073/pnas.2421...
-
[23]
and Ng, Madelena Y and Pannu, Jaspreet and Re, Christopher and Schmok, Jonathan C and St
Brixi, Garyk and Durrant, Matthew G and Ku, Jerome and Poli, Michael and Brockman, Greg and Chang, Daniel and Gonzalez, Gabriel A and King, Samuel H and Li, David B and Merchant, Aditi T and Naghipourfar, Mohsen and Nguyen, Eric and Ricci-Tam, Chiara and Romero, David W and Sun, Gwanggyu and Taghibakshi, Ali and Vorontsov, Anton and Yang, Brandon and Deng...
2025
-
[24]
2025 , doi =
Cheng, Wenduo and Song, Zhenqiao and Zhang, Yang and Wang, Shike and Wang, Danqing and Yang, Muyu and Li, Lei and Ma, Jian , title =. 2025 , doi =. https://www.biorxiv.org/content/early/2025/01/08/2025.01.06.631595.full.pdf , journal =
2025
-
[25]
2025 , eprint=
Gene42: Long-Range Genomic Foundation Model With Dense Attention , author=. 2025 , eprint=
2025
-
[26]
BiDNAMamba: Pre-trained Bidirectional State Space Model for Motif Analysis in DNA , doi =
Zeng, Guangjian and Yu, Xiaxia and Tao, Siyuan and Zhou, Weiye and Xiong, Momiao and Fang, Shenying , year =. BiDNAMamba: Pre-trained Bidirectional State Space Model for Motif Analysis in DNA , doi =
-
[27]
BioToken and BioFM
Medvedev, Aleksandr and Viswanathan, Karthik and Kanithi, Praveenkumar and Vishniakov, Kirill and Munjal, Prateek and Christophe, Cl. BioToken and BioFM. 2025 , doi =. https://www.biorxiv.org/content/early/2025/04/01/2025.03.27.645711.full.pdf , journal =
2025
-
[28]
2024 , doi =
Zhang, Xiang and Yang, Mingjie and Yin, Xunhang and Qian, Yining and Sun, Fei , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/04/28/2024.04.24.590879.full.pdf , journal =
2024
-
[29]
2024 , doi =
Ye, Peng and Bai, Weiqing and Ren, Yuchen and Li, Wenran and Qiao, Lifeng and Liang, Chaoqi and Wang, Linxiao and Cai, Yuchen and Sun, Jianle and Yang, Zejun and Zheng, Peng and Dong, Nanqing and Chen, Tao and Wang, Zhihui and Liu, Xihui and Ma, Xinzhu and Yan, Hongliang and Wang, Zhen and Wang, Sijia and Ouyang, Wanli , title =. 2024 , doi =. https://www...
2024
-
[30]
and Ye, Jieping and Li, Jun and Shu, Yuelong and Shi, Mang and Li, Zhaorong , title =
He, Yong and Fang, Pan and Shan, Yongtao and Pan, Yuanfei and Wei, Yanhong and Chen, Yichang and Chen, Yihao and Liu, Yi and Zeng, Zhenyu and Zhou, Zhan and Zhu, Feng and Holmes, Edward C. and Ye, Jieping and Li, Jun and Shu, Yuelong and Shi, Mang and Li, Zhaorong , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/05/14/2024.05.10.592927....
2024
-
[31]
2025 , doi =
Long, Weicai and Su, Houcheng and Xiong, Jiaqi and Zhang, Yanlin , title =. 2025 , doi =. https://www.biorxiv.org/content/early/2025/01/25/2025.01.23.634452.full.pdf , journal =
2025
-
[32]
2025 , eprint=
HybriDNA: A Hybrid Transformer-Mamba2 Long-Range DNA Language Model , author=. 2025 , eprint=
2025
-
[33]
2024 , eprint=
Model Decides How to Tokenize: Adaptive DNA Sequence Tokenization with MxDNA , author=. 2024 , eprint=
2024
-
[34]
2024 , eprint=
VQDNA: Unleashing the Power of Vector Quantization for Multi-Species Genomic Sequence Modeling , author=. 2024 , eprint=
2024
-
[35]
2025 , eprint=
BMFM-DNA: A SNP-aware DNA foundation model to capture variant effects , author=. 2025 , eprint=
2025
-
[36]
and Ku, Jerome and Poli, Michael and Brockman, Greg and Chang, Daniel and Gonzalez, Gabriel A
Brixi, Garyk and Durrant, Matthew G. and Ku, Jerome and Poli, Michael and Brockman, Greg and Chang, Daniel and Gonzalez, Gabriel A. and King, Samuel H. and Li, David B. and Merchant, Aditi T. and Naghipourfar, Mohsen and Nguyen, Eric and Ricci-Tam, Chiara and Romero, David W. and Sun, Gwanggyu and Taghibakshi, Ali and Vorontsov, Anton and Yang, Brandon an...
2025
-
[37]
and Dalla-Torre, Hugo and Blum, Christopher and Hexemer, Lorenz and Pandey, Priyanka and Laurent, Stefan and Lopez, Marie and Laterre, Alexandre and Lang, Maren and
Richard, Guillaume and de Almeida, Bernardo P. and Dalla-Torre, Hugo and Blum, Christopher and Hexemer, Lorenz and Pandey, Priyanka and Laurent, Stefan and Lopez, Marie and Laterre, Alexandre and Lang, Maren and. ChatNT: A Multimodal Conversational Agent for DNA, RNA and Protein Tasks , elocation-id =. 2024 , doi =. https://www.biorxiv.org/content/early/2...
2024
-
[38]
Boshar, Sam and Evans, Benjamin and Tang, Ziqi and Picard, Armand and Adel, Yanis and Lorbeer, Franziska K. and Rajesh, Chandana and Karch, Tristan and Sidbon, Shawn and Emms, David and Mendoza-Revilla, Javier and Al-Ani, Fatimah and Seitz, Evan and Schiff, Yair and Bornachot, Yohan and Hernandez, Ariana and Lopez, Marie and Laterre, Alexandre and Beguir,...
2025
-
[39]
and Ahanger, Sajad H
Mclaughlin, Shae M. and Ahanger, Sajad H. and Lim, Daniel A. , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/12/03/2024.11.27.625761.full.pdf , journal =
2024
-
[40]
2023 , doi =
Gao, Zijing and Liu, Qiao and Zeng, Wanwen and Wong, Wing Hung and Jiang, Rui , title =. 2023 , doi =. https://www.biorxiv.org/content/early/2023/07/18/2023.07.15.549134.full.pdf , journal =
2023
-
[41]
Malusare, Aditya and Kothandaraman, Harish and Tamboli, Dipesh and Lanman, Nadia A and Aggarwal, Vaneet , editor=. Understanding the natural language of DNA using encoder–decoder foundation models with byte-level precision , volume=. Bioinformatics Advances , publisher=. doi:10.1093/bioadv/vbae117 , number=
-
[42]
2025 , eprint=
HAD: Hybrid Architecture Distillation Outperforms Teacher in Genomic Sequence Modeling , author=. 2025 , eprint=
2025
-
[43]
C.La.P.: Enhancing transformer-based genomic signal modeling by integrating DNA sequences and chromatin accessibility data , elocation-id =
Nisantzis, Panos Firbas and Gon. C.La.P.: Enhancing transformer-based genomic signal modeling by integrating DNA sequences and chromatin accessibility data , elocation-id =. 2025 , doi =. https://www.biorxiv.org/content/early/2025/02/23/2025.02.19.638643.full.pdf , journal =
2025
-
[44]
2022 , doi =
Gresova, Katarina and Martinek, Vlastimil and Cechak, David and Simecek, Petr and Alexiou, Panagiotis , title =. 2022 , doi =. https://www.biorxiv.org/content/early/2022/06/10/2022.06.08.495248.full.pdf , journal =
2022
-
[45]
2025 , eprint=
OmniGenBench: A Modular Platform for Reproducible Genomic Foundation Models Benchmarking , author=. 2025 , eprint=
2025
-
[46]
2024 , doi =
Feng, Haonan and Wu, Lang and Zhao, Bingxin and Huff, Chad and Zhang, Jianjun and Wu, Jia and Lin, Lifeng and Wei, Peng and Wu, Chong , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/08/18/2024.08.16.608288.full.pdf , journal =
2024
-
[47]
2025 , eprint=
OmniGenBench: A Benchmark for Omnipotent Multimodal Generation across 50+ Tasks , author=. 2025 , eprint=
2025
-
[48]
bioRxiv preprint bioRxiv:2025.06.25.661622 , year=
Genomic Touchstone: Benchmarking Genomic Language Models in the Context of the Central Dogma , author=. bioRxiv preprint bioRxiv:2025.06.25.661622 , year=
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.