Closing the Prior-Posterior Loop: Self-Reflective Molecular Design with Analysis-Driven LLM Iteration
Pith reviewed 2026-06-27 14:36 UTC · model grok-4.3
The pith
Full first-principles outputs turn LLMs from trial-and-error samplers into causal molecular designers.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
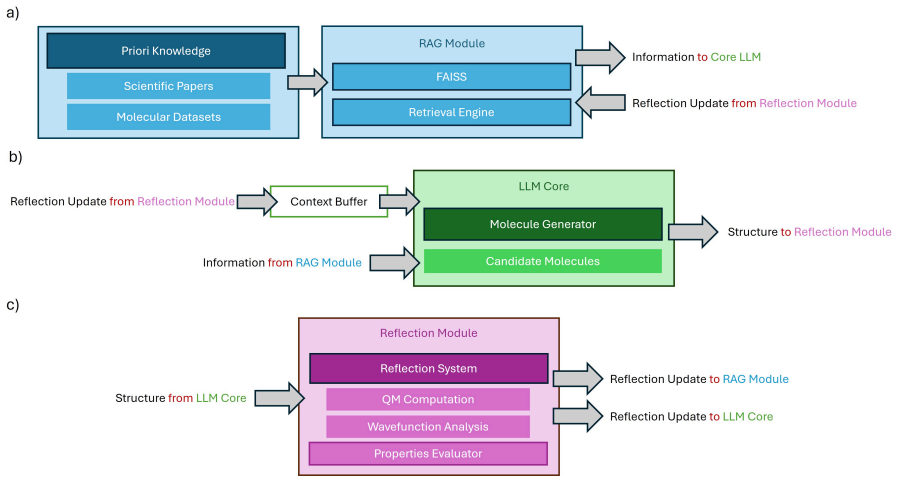
Coupling retrieval-augmented generation with a self-reflection module that feeds orbital energies, atomic charges, and electron densities from first-principles calculations back into the design loop transforms the LLM from a stochastic sampler into a causal reasoner that understands not only that a molecule fails but why.
What carries the argument
The SPR reflection module that ingests full physicochemical outputs to drive iterative structure-property reasoning inside the prior-posterior loop.
If this is right
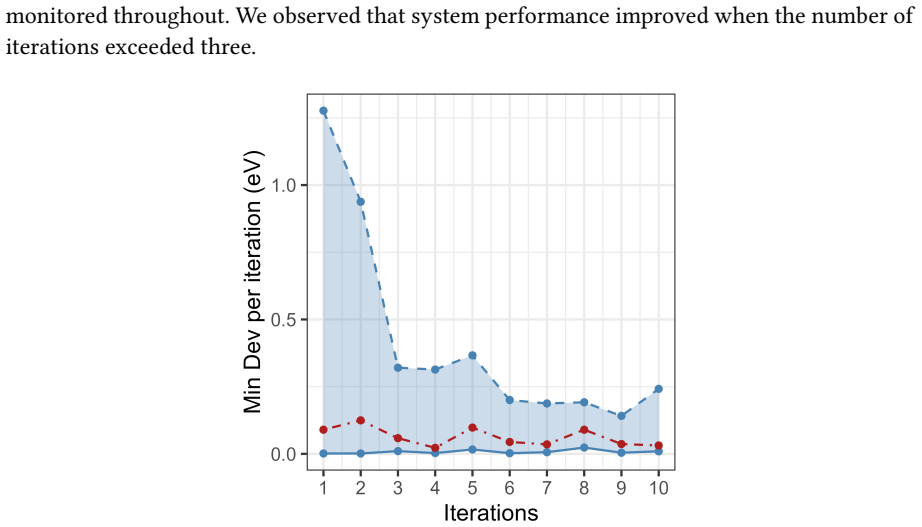
- Deviation reaches 0.0014 eV with 100 percent success rate on HOMO-LUMO targets from 2.0 to 5.0 eV under the SPR plus RAG configuration.
- The method consistently beats scalar-feedback and non-reflective baselines on both median and mean deviation.
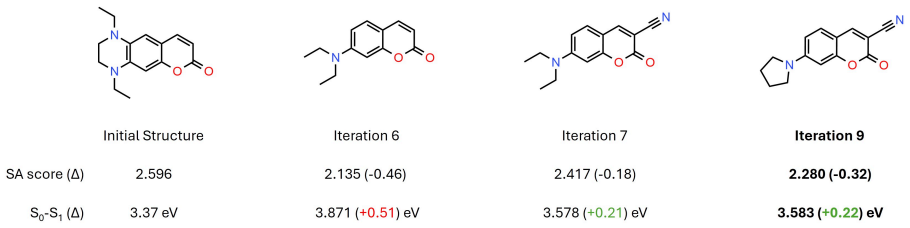
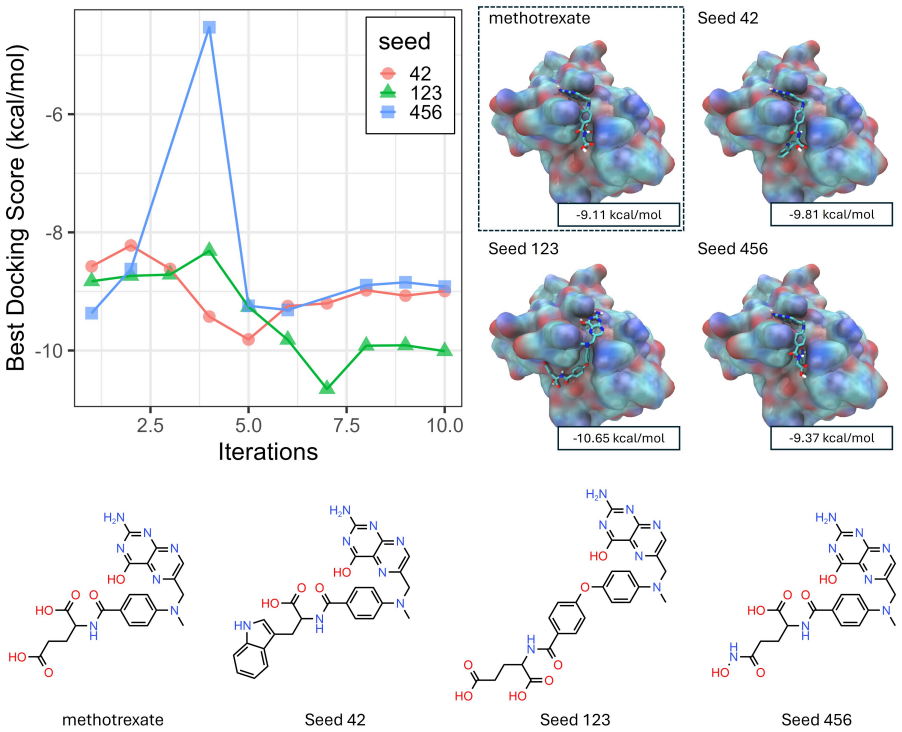
- The same framework generalizes directly to dipole-moment design, synthetic accessibility optimization, and molecular docking.
- Performance remains stable across seven distinct LLM backbones.
Where Pith is reading between the lines
- Richer feedback may cut the number of design cycles needed before experimental validation in early molecular discovery.
- The same reflection pattern could transfer to LLM-driven design in adjacent fields such as materials or catalyst screening.
- Future work could test whether the causal reasoning holds when first-principles data are replaced by cheaper surrogate models.
Load-bearing premise
That the LLM performs genuine causal reasoning from the provided first-principles data rather than improved pattern matching or prompting effects.
What would settle it
A controlled comparison in which an LLM given the complete orbital energies, charges, and densities still matches the performance of one given only the scalar HOMO-LUMO value on the same target set.
Figures




read the original abstract
Can a general-purpose large language model design molecules with the precision of a seasoned chemist? Current LLM-based frameworks answer this question with scalar feedback loops - generate, score, reject - that amount to informed trial-and-error. Here we show that replacing a single number with the full physicochemical rationale from first-principles calculations transforms the LLM from a stochastic sampler into a causal reasoner. Our system couples retrieval-augmented generation with a self-reflection module that feeds orbital energies, atomic charges, and electron densities - rather than compressed scores - back into the design loop. On HOMO-LUMO gap targets from 2.0 to 5.0 eV, this structure-property-relationship (SPR) reflection achieves a deviation as low as 0.0014 eV with a 100% success rate under the SPR+RAG configuration, consistently outperforming scalar-feedback and non-reflective baselines in median and mean deviation. The framework generalizes seamlessly to dipole-moment design, synthetic accessibility optimization, and molecular docking, and proves robust across 7 distinct LLM backbones. These results establish a new paradigm: when the model understands not only that a molecule fails, but why, iterative molecular design becomes genuinely mechanistic.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that feeding full first-principles outputs (orbital energies, atomic charges, electron densities) into an LLM via a self-reflection module within a retrieval-augmented generation loop transforms molecular design from scalar trial-and-error into causal reasoning. On HOMO-LUMO gap targets from 2.0 to 5.0 eV, the SPR+RAG configuration achieves deviations as low as 0.0014 eV with 100% success rate, outperforming scalar-feedback and non-reflective baselines in median/mean deviation; the framework is reported to generalize to dipole moment, synthetic accessibility, and docking tasks across 7 LLM backbones.
Significance. If the performance lift is reproducible and attributable to the detailed physicochemical inputs rather than format or prompting artifacts, the work could meaningfully advance LLM-driven molecular design by enabling iterative, rationale-based optimization. The reported robustness across backbones and extension to multiple properties would strengthen its practical value. The absence of controls for the causal-reasoning interpretation limits the strength of the central claim.
major comments (2)
- [Abstract] Abstract: the headline quantitative claim (0.0014 eV deviation, 100% success) is presented without any accompanying dataset description, error bars, number of trials, or statistical tests, rendering the result unverifiable from the provided information and load-bearing for all subsequent claims.
- [Abstract] Abstract (SPR reflection description): no ablation is described that substitutes the true first-principles outputs with scrambled, constant, or irrelevant numerical strings of matched length and format. Without this control, the attribution of performance gains to 'genuine causal reasoning' about structure-property relationships rather than richer prompting or retrieval effects cannot be assessed and directly underpins the paper's central mechanistic interpretation.
minor comments (2)
- [Abstract] Abstract: the generalization statements to dipole-moment design, synthetic accessibility, and docking lack any quantitative metrics or success criteria, weakening the breadth claim.
- [Abstract] Abstract: the distinction between 'SPR+RAG', 'scalar-feedback', and 'non-reflective' baselines is not defined at the level needed to interpret the reported outperformance.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which help strengthen the presentation and interpretation of our work. We address each major comment below and indicate the revisions we will make to the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the headline quantitative claim (0.0014 eV deviation, 100% success) is presented without any accompanying dataset description, error bars, number of trials, or statistical tests, rendering the result unverifiable from the provided information and load-bearing for all subsequent claims.
Authors: We agree that the abstract's headline claim would benefit from additional context to improve verifiability. In the revised version, we will modify the abstract to briefly describe the evaluation dataset (including the number of target molecules and the specific HOMO-LUMO gap range), report the number of trials or runs performed, include error bars or standard deviations where applicable, and reference the statistical methods used for comparison. These details are already present in the main text and supplementary information; we will ensure they are summarized in the abstract as well. revision: yes
-
Referee: [Abstract] Abstract (SPR reflection description): no ablation is described that substitutes the true first-principles outputs with scrambled, constant, or irrelevant numerical strings of matched length and format. Without this control, the attribution of performance gains to 'genuine causal reasoning' about structure-property relationships rather than richer prompting or retrieval effects cannot be assessed and directly underpins the paper's central mechanistic interpretation.
Authors: The referee raises a valid point regarding the need for a more direct control to isolate the contribution of meaningful physicochemical data. Our current experimental design includes comparisons to scalar-feedback baselines and non-reflective configurations, which help rule out simple prompting effects. However, to further address this, we will add an ablation study in the revised manuscript where the first-principles outputs are replaced with scrambled or constant values of matched length and format. This will allow us to quantify the specific benefit of the structured, meaningful data in the self-reflection module. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper describes an empirical LLM framework that feeds external first-principles outputs (orbital energies, atomic charges, electron densities) into a reflection module for molecular design. Reported metrics such as 0.0014 eV HOMO-LUMO deviation and 100% success rate are measured against independent quantum-chemistry targets rather than being defined by the method itself. No self-definitional equations, fitted parameters presented as predictions, load-bearing self-citations, or ansatzes smuggled via prior work appear in the derivation chain. The central claims remain falsifiable via external benchmarks and do not reduce to the inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
P. W. Anderson, More Is Different, Science 177, 393 (1972)
1972
-
[2]
Is Machine Learning Overhyped?, C&EN Global Enterprise 96, 16 (2018)
2018
-
[3]
Z. Qiao, A. S. Christensen, M. Welborn, F. R. Manby, A. Anandkumar, and T. F. Miller, Informing Geometric Deep Learning with Electronic Interactions to Accelerate Quantum Chemistry, Proceedings of the National Academy of Sciences 119, (2022)
2022
-
[4]
K. T. Butler, F. Oviedo, and P. Canepa, Machine Learning in Materials Science (American Chemical Society, 2021)
2021
-
[5]
D. B. Catacutan, J. Alexander, A. Arnold, and J. M. Stokes, Machine Learning in Preclin- ical Drug Discovery, Nature Chemical Biology 20, 960 (2024)
2024
-
[6]
J. L. McDonagh, N. Nath, L. De Ferrari, T. van Mourik, and J. B. O. Mitchell, Uniting Cheminformatics and Chemical Theory To Predict the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules, Journal of Chemical Information and Modeling 54, 844 (2014)
2014
-
[7]
S. Xia, E. Chen, and Y. Zhang, Integrated Molecular Modeling and Machine Learning for Drug Design, Journal of Chemical Theory and Computation 19, 7478 (2023)
2023
-
[8]
Javid, A
S. Javid, A. Rahmanulla, M. G. Ahmed, R. sultana, and B. R. Prashantha Kumar, Machine Learning & Deep Learning Tools in Pharmaceutical Sciences: A Comprehensive Review, Intelligent Pharmacy 3, 167 (2025)
2025
-
[9]
Kourou, T
K. Kourou, T. P. Exarchos, K. P. Exarchos, M. V. Karamouzis, and D. I. Fotiadis, Machine Learning Applications in Cancer Prognosis and Prediction, Computational and Struc - tural Biotechnology Journal 13, 8 (2015)
2015
-
[10]
Sanchez-Lengeling and A
B. Sanchez-Lengeling and A. Aspuru-Guzik, Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering, Science 361, 360 (2018)
2018
-
[11]
Y. Wang, Z. Li, and A. B. Farimani, Graph Neural Networks for Molecules , https://doi.org/ 10.48550/arXiv.2209.05582
-
[12]
Y. J. Lee, H. Kahng, and S. B. Kim, Generative Adversarial Networks for De Novo Molecular Design, Molecular Informatics 40, 2100045 (2021)
2021
-
[13]
DenseSteer: Steering Small Language Models towards Dense Math Reasoning
Y. Ouyang, S. Lin, and J.-E. Kim, Densesteer: Steering Small Language Models Towards Dense Math Reasoning , https://doi.org/10.48550/arXiv.2605.29247
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2605.29247
-
[14]
H. Ye et al., Evaluation-Driven Scaling for Scientific Discovery , https://doi.org/10.48550/ arXiv.2604.19341
-
[15]
N. Sothanaphan, Resolution of Erdős Problem #728: A Writeup of Aristotle's Lean Proof , https://doi.org/10.48550/arXiv.2601.07421
-
[16]
S. Liu, J. Wang, Y. Yang, C. Wang, L. Liu, H. Guo, and C. Xiao, Conversational Drug Editing Using Retrieval and Domain Feedback , in The Twelfth International Conference on Learning Representations, ICLR 2024, Vienna, Austria, May 7-11, 2024 (OpenReview.net, 2024)
2024
-
[17]
X. Nan, X. You, X. Liu, H. Liu, C. Ji, Y. Du, and J. Song, TaLiRAGen: Target-Aware Ligand Generation via Retrieval-Augmented Large Language Models, Molecular Diversity 30, 2699 (2026)
2026
-
[18]
S. Ito, K. Muraoka, and A. Nakayama, Knowledge-Informed Molecular Design for Zeolite Synthesis Using General-Purpose Pretrained Large Language Models Toward Human- Machine Collaboration, Chemistry of Materials 37, 2447 (2025)
2025
-
[19]
Zhang, X
P. Zhang, X. Peng, R. Han, T. Chen, and J. Ma, Rag2Mol: Structure-Based Drug Design Based on Retrieval Augmented Generation, Briefings in Bioinformatics 26, bbaf265 (2025)
2025
-
[20]
Stewart and M
I. Stewart and M. J. Buehler, Molecular Analysis and Design Using Generative Artificial Intelligence via Multi-Agent Modeling, Molecular Systems Design & Engineering 10, 314 (2025)
2025
-
[21]
Z. Hu, Y. Zhou, Z. Wang, X. Li, W. Yang, H. Fan, and Y. Yang, OSDA Agent: Leveraging Large Language Models for De Novo Design of Organic Structure Directing Agents , in (2024)
2024
-
[22]
Bhattacharya, H
D. Bhattacharya, H. J. Cassady, M. A. Hickner, and W. F. Reinhart, Large Language Models as Molecular Design Engines, Journal of Chemical Information and Modeling 64, 7086 (2024)
2024
-
[23]
K. D. Vogiatzis, Design of CO2-Philic Molecular Units with Large Language Models, Chemical Communications 61, 10166 (2025)
2025
-
[24]
K. T. Schütt, P.-J. Kindermans, H. E. Sauceda, S. Chmiela, A. Tkatchenko, and K.-R. Müller, Schnet: A Continuous-Filter Convolutional Neural Network for Modeling Quantum Interactions , https://doi.org/10.48550/arXiv.1706.08566
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1706.08566
-
[25]
J. Gasteiger, J. Groß, and S. Günnemann, Directional Message Passing for Molecular Graphs , https://doi.org/10.48550/arXiv.2003.03123
-
[26]
N. W. A. Gebauer, M. Gastegger, S. S. P. Hessmann, K.-R. Müller, and K. T. Schütt, Inverse Design of 3d Molecular Structures with Conditional Generative Neural Networks, Nature Communications 13, 973 (2022)
2022
-
[27]
T. Han, D. Yan, Q. Wu, N. Song, H. Zhang, and D. Wang, Aggregation-Induced Emission: A Rising Star in Chemistry and Materials Science, Chinese Journal of Chemistry 39, 677 (2021)
2021
-
[28]
J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam, and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chemical Reviews 115, 11718 (2015)
2015
-
[29]
Pillai, A
O. Pillai, A. B. Dhanikula, and R. Panchagnula, Drug Delivery: An Odyssey of 100 Years, Current Opinion in Chemical Biology 5, 439 (2001)
2001
-
[30]
Ertl and A
P. Ertl and A. Schuffenhauer, Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions, Journal of Cheminformatics 1, 8 (2009)
2009
-
[31]
A. R. Jagtap, V. S. Satam, R. N. Rajule, and V. R. Kanetkar, The Synthesis and Characteri- zation of Novel Coumarin Dyes Derived from 1,4-Diethyl-1,2,3,4-Tetrahydro-7-Hydrox- yquinoxalin-6-Carboxaldehyde, Dyes and Pigments 82, 84 (2009)
2009
-
[32]
Neese, The ORCA Program System, Wires Computational Molecular Science 2, 73 (2012)
F. Neese, The ORCA Program System, Wires Computational Molecular Science 2, 73 (2012)
2012
-
[33]
Neese, Software Update: The \textsc{ORCA} Program System—Version 6.0, Wires Computational Molecular Science 15, e70019 (2025)
F. Neese, Software Update: The \textsc{ORCA} Program System—Version 6.0, Wires Computational Molecular Science 15, e70019 (2025)
2025
-
[34]
Neese, Software Update: The ORCA Program System—Version 5.0, Wires Computa - tional Molecular Science 12, e1606 (2022)
F. Neese, Software Update: The ORCA Program System—Version 5.0, Wires Computa - tional Molecular Science 12, e1606 (2022)
2022
-
[35]
D. R. Koes, M. P. Baumgartner, and C. J. Camacho, Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise, Journal of Chemical Informa - tion and Modeling 53, 1893 (2013)
2011
-
[36]
Quiroga and M
R. Quiroga and M. A. Villarreal, Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening, PLOS ONE 11, e155183 (2016)
2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.