Ab initio parametrization of distributed polarizable force fields
Pith reviewed 2026-06-27 15:41 UTC · model grok-4.3
The pith
Assigning polarizability to individual atoms as tensors rather than atom-type scalars improves force-field accuracy for ions and excited states.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Atomic polarizability tensors assigned to individual atoms, obtained from ab initio calculations on small organic molecules and predicted by a message-passing graph neural network, produce distributed polarizable force fields that describe neutral molecules more accurately and extend reliably to cations, anions, and excited states, all without adding cost to the molecular-dynamics step.
What carries the argument
Message-passing graph neural network that learns to map molecular graphs to per-atom polarizability tensors and scalars derived from first-principles calculations.
If this is right
- Force fields can now be used for simulations involving charged species and photo-excited molecules without separate parametrization schemes.
- Electronic response properties such as refractive index become accessible in large-scale molecular-dynamics runs at classical cost.
- A diagnostic based on the difference between predicted and reference polarizabilities flags molecules where the model is likely to fail.
- The same workflow applies to any conjugated organic building block once the network is trained on a representative set of small molecules.
Where Pith is reading between the lines
- The per-atom tensor approach may reduce the need for manual atom-type assignment when building models for new materials libraries.
- Because the network adds no runtime cost, the method could be inserted into existing polarizable simulation packages with minimal code changes.
- Extending the training set to include inorganic ions or metal-organic complexes would test whether the same transferability holds beyond organic molecules.
Load-bearing premise
Polarizability values calculated on small molecules can be transferred by the neural network to larger or charged molecules without introducing large systematic errors.
What would settle it
Direct comparison of the neural-network-predicted polarizabilities against new ab initio calculations for a molecule outside the training set that contains an ion or an excited-state geometry, showing whether the error remains below the threshold needed for accurate refractive-index or density-of-states predictions.
Figures
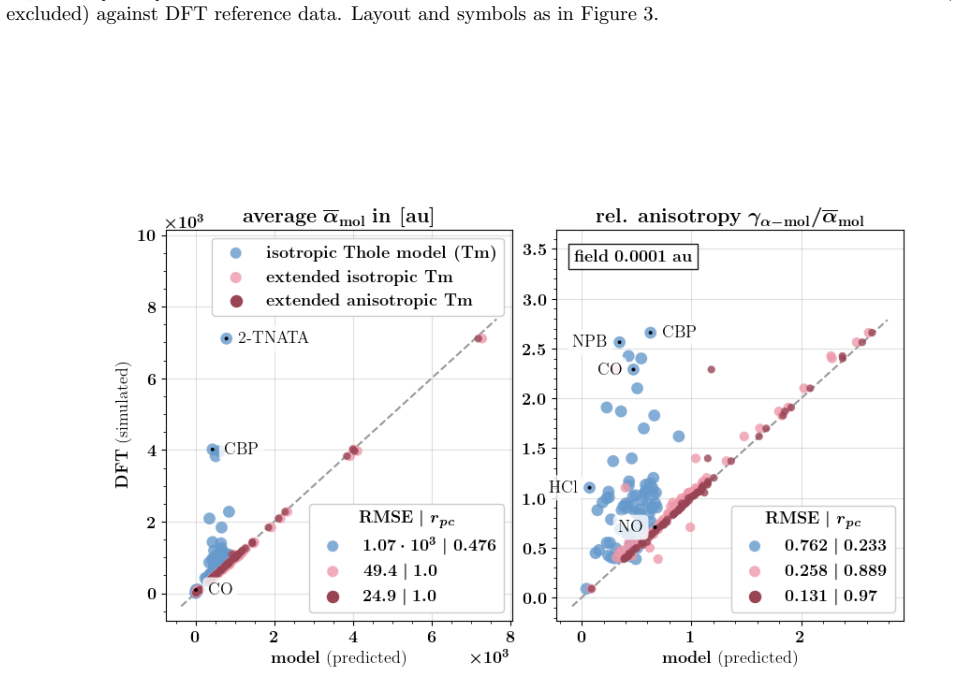
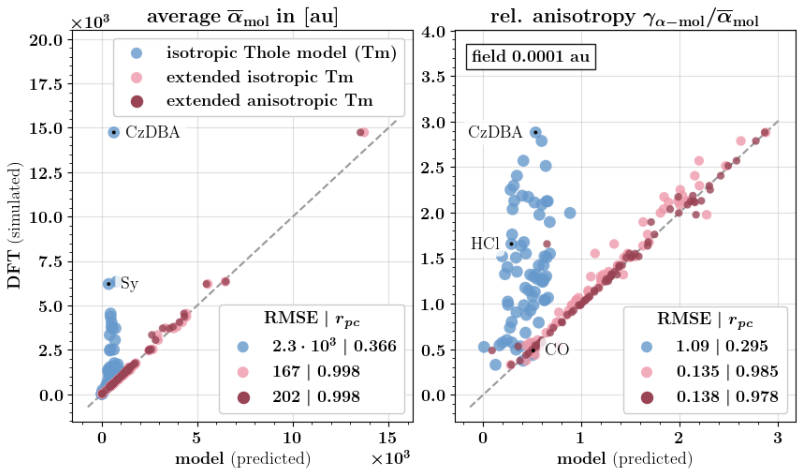
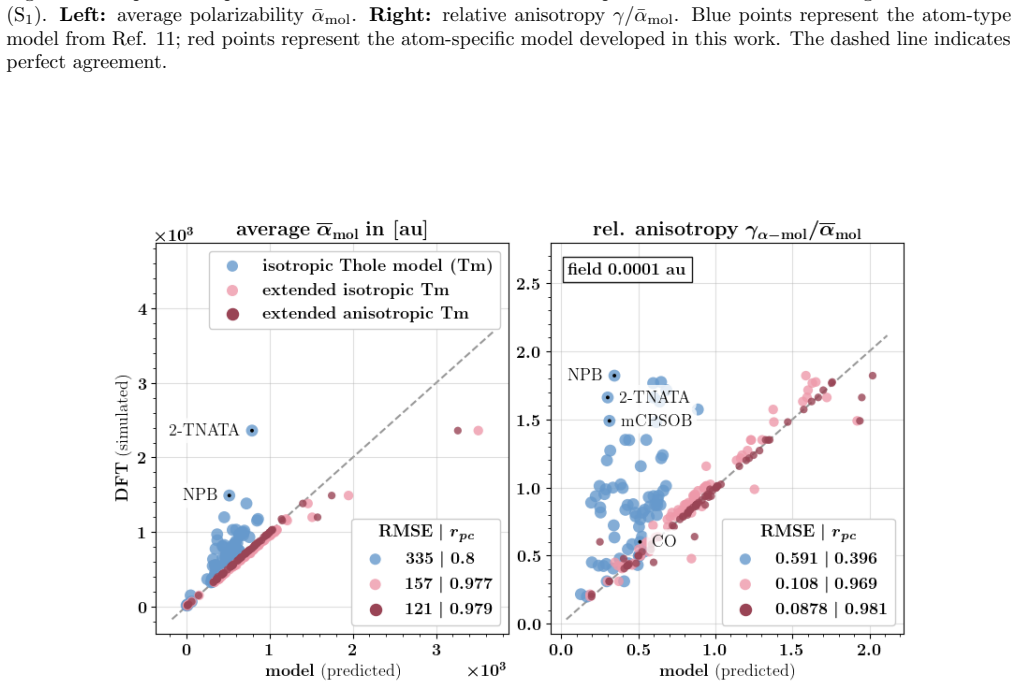
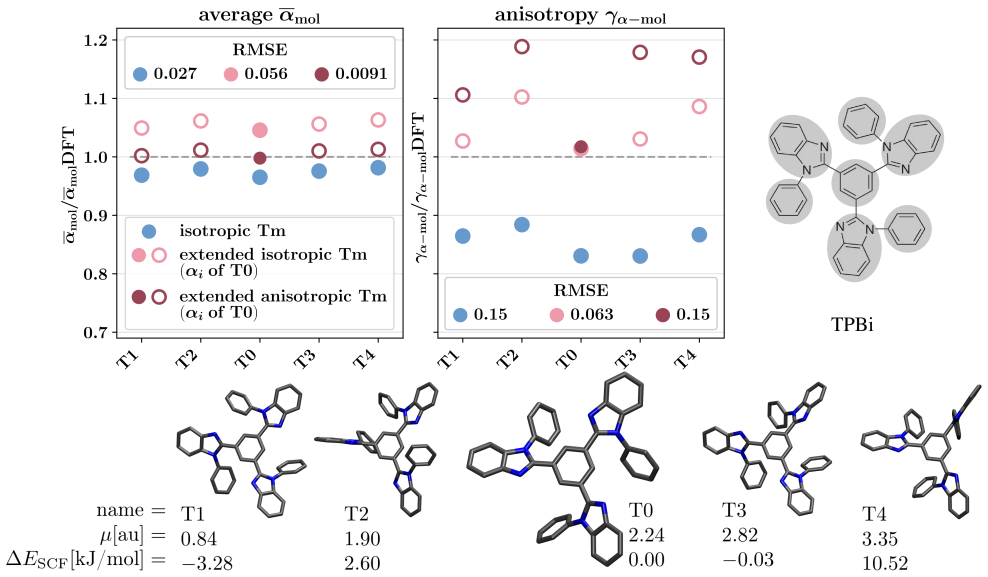
read the original abstract
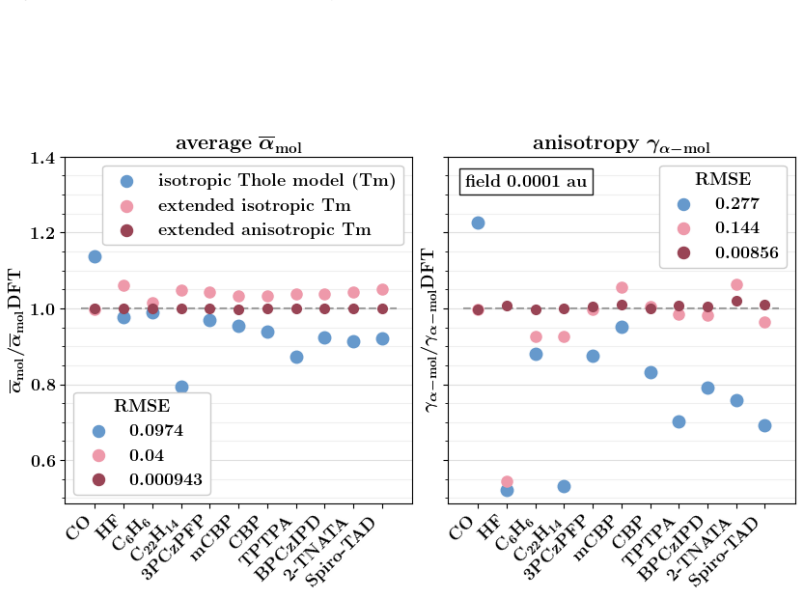
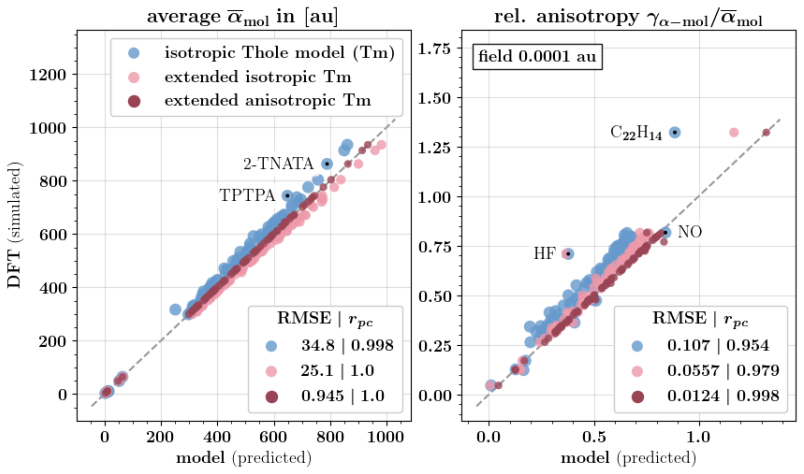
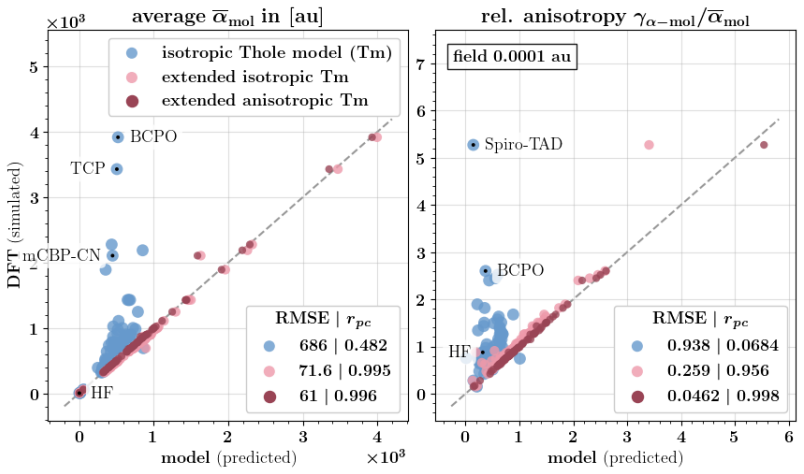
Polarizable force fields offer superior transferability and accuracy compared to classical force fields, enabling access to electronic response properties such as refractive index and electronic density of states. Here, we demonstrate two key improvements that significantly enhance their accuracy: (1) assigning atomic polarizability to individual atoms rather than atom types, and (2) employing atomic polarizability tensors instead of scalar values. These modifications extend the applicability of polarizable force fields to cations, anions, and excited states, while also providing more accurate descriptions of neutral molecules. We propose a first-principles-based parameterization procedure for atomic polarizability tensors and scalars, validated on a set of small organic molecules with conjugated building blocks. To overcome the computational cost of ab initio calculations, we train a message-passing graph neural network to predict polarizability parameters, enabling efficient and scalable parameterization. Crucially, this approach imposes no additional computational cost during simulations and provides a clear diagnostic criterion for identifying cases where polarizable force field models fail to accurately describe molecular polarizability.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that assigning per-atom polarizability tensors (rather than atom-type scalars) derived from ab initio calculations on small neutral organic molecules, then predicted via a message-passing GNN, yields more accurate distributed polarizable force fields with no extra simulation cost; this is asserted to extend applicability to cations, anions, and excited states while improving neutral-molecule descriptions, with a diagnostic for model failure.
Significance. If the transferability claims hold, the work would advance first-principles parametrization of polarizable force fields by replacing atom-type approximations with atom-specific tensors and enabling scalable GNN-based assignment; the absence of added computational cost during MD and the diagnostic criterion are concrete strengths.
major comments (2)
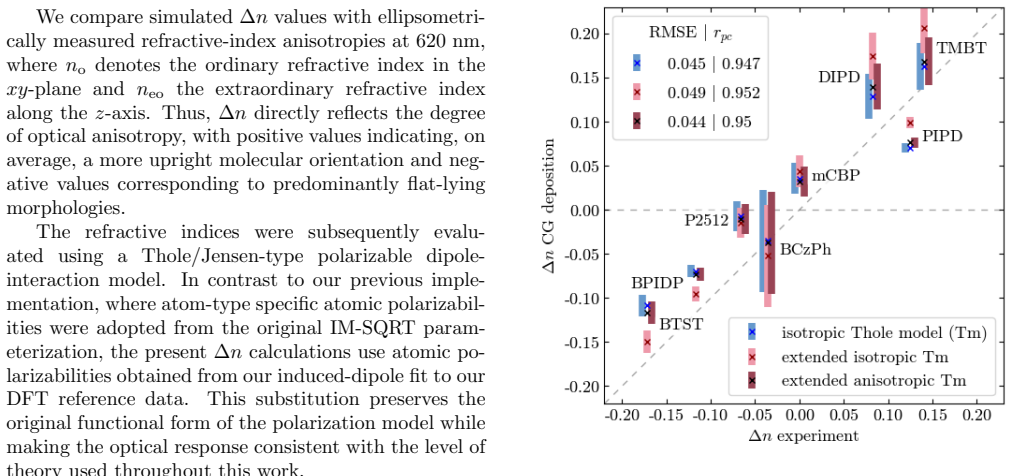
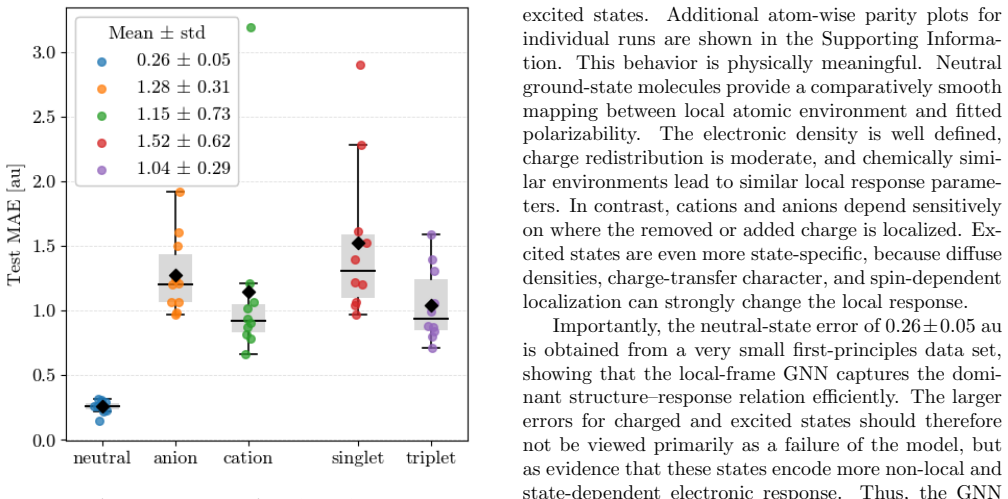
- [Abstract and §4] Abstract and §4 (validation): the central claim that the per-atom tensor approach 'extends the applicability ... to cations, anions, and excited states' is unsupported because the reported ab initio reference data and GNN predictions are confined to neutral ground-state molecules with conjugated blocks; no separate calculations or transfer tests for charged or electronically excited systems are provided, leaving the extension as an assumption rather than a demonstrated result.
- [§3] §3 (GNN training): while the GNN is trained on ab initio polarizabilities from small molecules, the manuscript does not report error metrics or transferability tests on systems outside the neutral training distribution, which is required to substantiate the broader applicability asserted in the abstract.
minor comments (2)
- [§2] Notation for the polarizability tensor components should be clarified with an explicit equation relating the distributed atomic tensors to the molecular polarizability.
- [Figure 2] Figure captions for the GNN architecture and validation plots should include the exact training/validation split sizes and the molecules used.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We agree that the claims regarding extension to cations, anions, and excited states are not supported by explicit calculations in the current work and must be revised to reflect the demonstrated scope on neutral ground-state molecules. We will update the abstract, §3, and §4 accordingly while preserving the core technical contributions on per-atom tensors and the GNN predictor.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (validation): the central claim that the per-atom tensor approach 'extends the applicability ... to cations, anions, and excited states' is unsupported because the reported ab initio reference data and GNN predictions are confined to neutral ground-state molecules with conjugated blocks; no separate calculations or transfer tests for charged or electronically excited systems are provided, leaving the extension as an assumption rather than a demonstrated result.
Authors: We acknowledge that the manuscript provides no ab initio data or GNN predictions for charged or excited systems, so the extension claim is prospective rather than demonstrated. The per-atom tensor formulation is motivated by the expectation that local electronic response (captured via ab initio tensors) will adapt to changes in charge or electronic state where fixed atom-type scalars cannot, but this remains an untested hypothesis here. We will revise the abstract and §4 to state that the approach is designed to enable such extensions and that explicit validation on cations, anions, and excited states is planned for future work. revision: yes
-
Referee: [§3] §3 (GNN training): while the GNN is trained on ab initio polarizabilities from small molecules, the manuscript does not report error metrics or transferability tests on systems outside the neutral training distribution, which is required to substantiate the broader applicability asserted in the abstract.
Authors: The GNN is trained and evaluated exclusively on the neutral small-molecule dataset; no error metrics or tests on out-of-distribution systems (charged species, excited states, or larger molecules) are reported. We agree this limits substantiation of the broader applicability stated in the abstract. We will revise §3 to explicitly delimit the current training distribution, add a discussion of expected limitations for out-of-distribution cases, and reference the diagnostic criterion already in the manuscript as a practical safeguard for new systems. The abstract will be updated to match. revision: yes
Circularity Check
No significant circularity; derivation uses external ab initio references
full rationale
The parameterization procedure starts from ab initio calculations on small neutral organic molecules to obtain per-atom polarizability tensors and scalars, then trains a message-passing GNN on those derived values to enable prediction for new molecules. No step reduces a claimed prediction or first-principles result to its own inputs by construction, nor does any load-bearing premise rest on a self-citation chain. The extension to cations, anions, and excited states is asserted on the basis of the per-atom tensor representation but is not validated within the described neutral-molecule test set; this constitutes an untested transferability assumption rather than circularity. The overall chain remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- GNN model parameters and hyperparameters
axioms (1)
- domain assumption Ab initio quantum calculations on small organic molecules provide transferable reference values for atomic polarizability tensors and scalars
Reference graph
Works this paper leans on
-
[2]
Physical Chemistry Chemical Physics , author =
Using atomic charges to model molecular polarization , volume =. Physical Chemistry Chemical Physics , author =. 2022 , note =. doi:10.1039/D1CP03542H , abstract =
-
[3]
The Journal of Physical Chemistry A , author =
Ambiguities in. The Journal of Physical Chemistry A , author =. 2024 , note =. doi:10.1021/acs.jpca.4c01890 , abstract =
-
[4]
Journal of Chemical Theory and Computation , author =
Unifying. Journal of Chemical Theory and Computation , author =. 2023 , note =. doi:10.1021/acs.jctc.3c00341 , abstract =
-
[5]
The journal of physical chemistry
A. The journal of physical chemistry. A , author =. 2009 , pmid =. doi:10.1021/jp808952m , abstract =
-
[6]
Chemical Physics Letters , author =. 2007 , keywords =. doi:10.1016/j.cplett.2007.02.065 , abstract =
-
[8]
Annual Review of Biophysics , author =
Polarizable. Annual Review of Biophysics , author =. 2019 , keywords =. doi:10.1146/annurev-biophys-070317-033349 , abstract =
-
[9]
Advanced Energy Materials , volume =
Scherer, Christoph and Kinaret, Naomi and Lin, Kun-Han and Qaisrani, Muhammad Nawaz and Post, Felix and May, Falk and Andrienko, Denis , title =. Advanced Energy Materials , volume =. doi:https://doi.org/10.1002/aenm.202403124 , url =. https://advanced.onlinelibrary.wiley.com/doi/pdf/10.1002/aenm.202403124 , year =
-
[10]
M. J. Frisch and G. W. Trucks and H. B. Schlegel and G. E. Scuseria and M. A. Robb and J. R. Cheeseman and G. Scalmani and V. Barone and G. A. Petersson and H. Nakatsuji and X. Li and M. Caricato and A. V. Marenich and J. Bloino and B. G. Janesko and R. Gomperts and B. Mennucci and H. P. Hratchian and J. V. Ortiz and A. F. Izmaylov and J. L. Sonnenberg an...
2016
-
[11]
2026 , eprint=
Vibrational infrared and Raman spectra of the methanol molecule with equivariant neural-network property surfaces , author=. 2026 , eprint=
2026
-
[12]
Derek A. Long , title =. 2002 , publisher =. doi:10.1002/0470845767 , URL =
-
[13]
Zeitschrift für Physik , volume =
George Placzek , title =. Zeitschrift für Physik , volume =. 1931 , doi =
1931
-
[14]
Peach, Michael J. G. and Williamson, Matthew J. and Tozer, David J. , title =. Journal of Chemical Theory and Computation , volume =. 2011 , doi =
2011
-
[15]
The Journal of Chemical Physics , volume =
Dreuw, Andreas and Weisman, Jennifer L. and Head-Gordon, Martin , title =. The Journal of Chemical Physics , volume =. 2003 , month =. doi:10.1063/1.1590951 , url =
-
[16]
Physical Chemistry Chemical Physics , author =
Evaluating excited state atomic polarizabilities of chromophores , volume =. Physical Chemistry Chemical Physics , author =. 2018 , keywords =. doi:10.1039/C7CP08549D , number =
-
[17]
The Journal of Chemical Physics , author =
Polarizability of molecular clusters as calculated by a dipole interaction model , volume =. The Journal of Chemical Physics , author =. 2002 , pages =. doi:10.1063/1.1433747 , language =
-
[18]
Journal of Chemical Theory and Computation , volume =
Liang, Jiashu and Feng, Xintian and Hait, Diptarka and Head-Gordon, Martin , title =. Journal of Chemical Theory and Computation , volume =. 2022 , doi =
2022
-
[19]
and Limas, Nidia Gabaldon , title =
Manz, Thomas A. and Limas, Nidia Gabaldon , title =. RSC Adv. , year =. doi:10.1039/C6RA04656H , url =
-
[20]
Limas, Nidia Gabaldon and Manz, Thomas A. , title =. RSC Adv. , year =. doi:10.1039/C6RA05507A , url =
-
[21]
Limas, Nidia Gabaldon and Manz, Thomas A. , title =. RSC Adv. , year =. doi:10.1039/C7RA11829E , url =
-
[22]
Martin H. Müser and Sergey V. Sukhomlinov and Lars Pastewka , title =. Advances in Physics: X , volume =. 2023 , publisher =. doi:10.1080/23746149.2022.2093129 , url =
-
[23]
Journal of Chemical Theory and Computation , author =
Polarizable. Journal of Chemical Theory and Computation , author =. 2011 , pages =. doi:10.1021/ct200304d , language =
-
[24]
Versatile Object-Oriented Toolkit for Coarse-Graining Applications , journal =
R. Versatile Object-Oriented Toolkit for Coarse-Graining Applications , journal =. 2009 , doi =
2009
-
[25]
Molecular polarizabilities calculated with a modified dipole interaction , journal =. 1981 , issn =. doi:https://doi.org/10.1016/0301-0104(81)85176-2 , url =
-
[26]
1971 , journal =
Iterative solution of large linear systems , author =. 1971 , journal =
1971
-
[27]
Atom dipole interaction model for molecular polarizability
Applequist, Jon and Carl, James R and Fung, Kwok-Kueng , year =. Atom dipole interaction model for molecular polarizability. Journal of the American Chemical Society , number =
-
[28]
The Journal of Chemical Physics , author =
Excited state polarizabilities of conjugated molecules calculated using time dependent density functional theory , volume =. The Journal of Chemical Physics , author =. 2001 , pages =. doi:10.1063/1.1415085 , abstract =
-
[29]
and Liu, Shubin , month = jan, year =
Zhao, Dongbo and He, Xin and Ayers, Paul W. and Liu, Shubin , month = jan, year =. Excited-. Molecules , publisher =. doi:10.3390/molecules28062576 , abstract =
-
[30]
Ayers, Paul W. , month = dec, year =. The physical basis of the hard/soft acid/base principle , volume =. Faraday Discussions , publisher =. doi:10.1039/B606877D , abstract =
-
[31]
International conference on machine learning , pages=
E (n) equivariant graph neural networks , author=. International conference on machine learning , pages=. 2021 , organization=
2021
-
[32]
arXiv preprint arXiv:2511.07087 , year=
Direct Molecular Polarizability Prediction with SO (3) Equivariant Local Frame GNNs , author=. arXiv preprint arXiv:2511.07087 , year=
-
[33]
arXiv preprint arXiv:2405.15389 , year=
Beyond canonicalization: How tensorial messages improve equivariant message passing , author=. arXiv preprint arXiv:2405.15389 , year=
-
[34]
Journal of Chemical Theory and Computation , volume=
Kernel-based minimal distributed charges: A conformationally dependent ESP-model for molecular simulations , author=. Journal of Chemical Theory and Computation , volume=. 2024 , publisher=
2024
-
[35]
Journal of Chemical Theory and Computation , volume=
Molecular dynamics with conformationally dependent, distributed charges , author=. Journal of Chemical Theory and Computation , volume=. 2022 , publisher=
2022
-
[36]
Journal of chemical theory and computation , volume=
Polarizable multipolar molecular dynamics using distributed point charges , author=. Journal of chemical theory and computation , volume=. 2020 , publisher=
2020
-
[37]
, title =
Stone, Anthony J. , title =. Journal of Chemical Theory and Computation , year =
-
[38]
Physical Chemistry Chemical Physics , year =
Pracht, Philipp and Bohle, Fabian and Grimme, Stefan , title =. Physical Chemistry Chemical Physics , year =
-
[39]
Journal of Chemical Theory and Computation , year =
Bannwarth, Christoph and Ehlert, Sebastian and Grimme, Stefan , title =. Journal of Chemical Theory and Computation , year =
-
[40]
Acta Crystallographica Section A , year =
Kabsch, Wolfgang , title =. Acta Crystallographica Section A , year =
-
[41]
Chaudhry, Imran and Bronson, Mark J. Jr. and Jensen, Lasse , month = jul, year =. -. Journal of Chemical Theory and Computation , publisher =. doi:10.1021/acs.jctc.5c00752 , number =
-
[42]
Molecular Dynamics Simulations Based on Polarizable Models Show that Ion Permeation Interconverts between Different Mechanisms as a Function of Membrane Thickness , journal =
Chen, Peiran and Vorobyov, Igor and Roux, Beno. Molecular Dynamics Simulations Based on Polarizable Models Show that Ion Permeation Interconverts between Different Mechanisms as a Function of Membrane Thickness , journal =. 2021 , volume =
2021
-
[43]
Analyzing Dynamical Disorder for Charge Transport in Organic Semiconductors via Machine Learning , journal =
Reiser, Patrick and Konrad, Manuel and Fediai, Artem and Le. Analyzing Dynamical Disorder for Charge Transport in Organic Semiconductors via Machine Learning , journal =. 2021 , volume =
2021
-
[44]
and Papageorgiou, D
Kaklamanis, K. and Papageorgiou, D. G. , title =. Computer Physics Communications , year =
-
[45]
and Fu, Yao-Tsung and Risko, Chad and Br
Ryno, Sean M. and Fu, Yao-Tsung and Risko, Chad and Br. Polarization Energies at Organic--Organic Interfaces: Impact on the Charge Separation Barrier at Donor--Acceptor Interfaces in Organic Solar Cells , journal =. 2016 , volume =
2016
-
[46]
The Journal of Physical Chemistry A , author =
Molecular and. The Journal of Physical Chemistry A , author =. 1998 , keywords =. doi:10.1021/jp980221f , language =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.