Can DFT-trained neural network potentials reproduce structure, solvation, and water-exchange properties in aqueous magnesium solutions?
Pith reviewed 2026-06-30 11:21 UTC · model grok-4.3
The pith
DFT-trained neural network potentials reproduce magnesium ion hydration structure, diffusion, ion pairing, and water exchange kinetics but underestimate solvation free energies.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
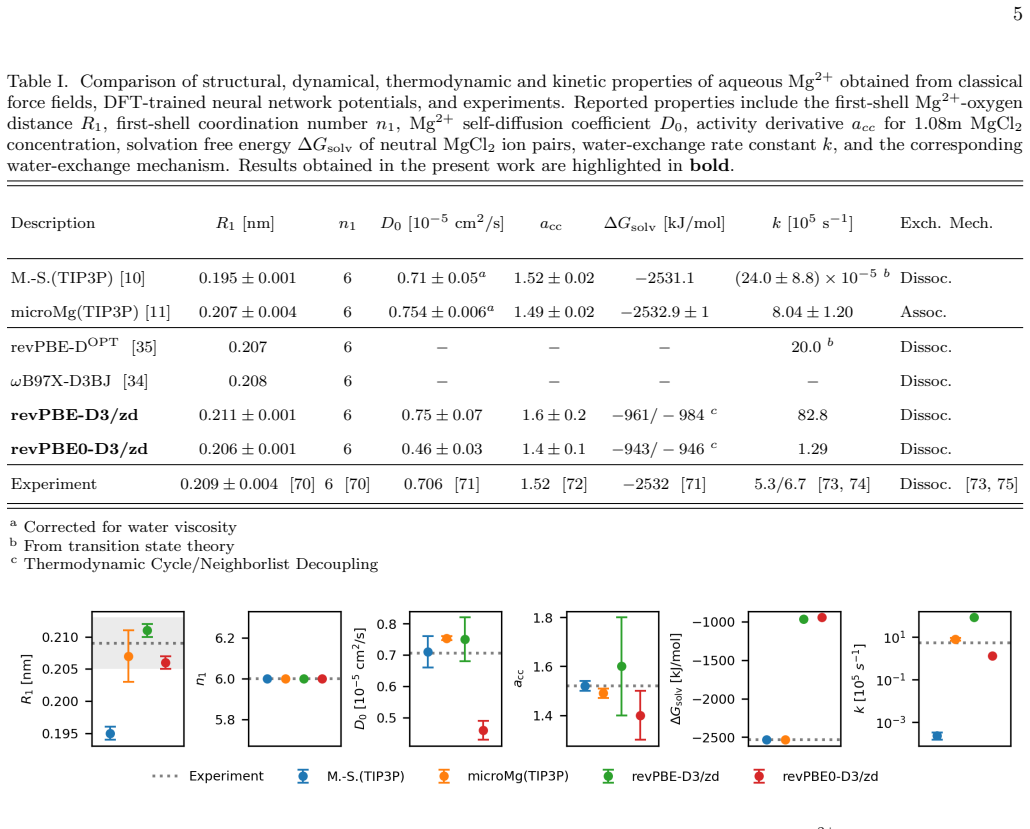
MACE neural network potentials trained on revPBE-D3/zd and revPBE0-D3/zd reference data reproduce the octahedral structure of the first hydration shell of Mg2+, diffusion coefficients, ion pairing properties, and water-exchange rates within one order of magnitude of experiment when combined with transition interface sampling, which reveals a dissociative exchange mechanism. The NNP-derived solvation free energy significantly underestimates the experimental value, revealing a limitation of the present local NNP architectures for describing ion solvation thermodynamics and the need for explicit long-range electrostatic treatments.
What carries the argument
MACE neural network potentials trained on DFT data for aqueous MgCl2, paired with transition path sampling and transition interface sampling to access rare water-exchange events.
If this is right
- The first hydration shell of Mg2+ remains octahedral and matches experimental structure.
- Diffusion coefficients of the ions agree with measured values.
- Water exchange follows a dissociative mechanism at rates within one order of magnitude of experiment.
- Ion pairing properties are described accurately.
- Solvation free energies require explicit long-range electrostatic treatments for quantitative agreement with experiment.
Where Pith is reading between the lines
- These NNPs could be used in larger biomolecular simulations to study magnesium-dependent enzymes or nucleic acids.
- The same training approach might resolve similar kinetic and structural shortcomings for other divalent cations.
- Testing whether long-range electrostatic corrections preserve the good performance on structure and kinetics would be a direct next step.
- The results suggest machine-learned potentials can outperform classical fields on rare-event ion kinetics in solution.
Load-bearing premise
Local neural network architectures trained on the chosen DFT data capture all relevant many-body effects for the tested properties except solvation thermodynamics.
What would settle it
Adding an explicit long-range electrostatic correction to the trained NNP, recomputing the solvation free energy, and checking whether the result then matches the experimental value for Mg2+.
Figures
read the original abstract
Magnesium ions play an essential role in many biological processes but remain challenging to model in biomolecular simulations. Despite considerable scientific effort, classical force fields fail to simultaneously reproduce key structural, thermodynamic and kinetic solution properties, likely due to their inability to explicitly account for quantum many-body effects. Here, we develop and systematically benchmark MACE neural network potentials (NNPs) for aqueous MgCl$_2$ solutions trained on revPBE-D3/zd and revPBE0-D3/zd density functional theory reference data and assess their ability to reproduce a broad range of experimental solution properties including the structure of the first hydration shell, diffusion coefficient, activity derivative, water-exchange rate and mechanism as well as solvation free energy. Both NNPs accurately reproduce the octahedral structure of the first hydration shell, ion pairing properties and diffusion coefficients. Combining the NNPs with transition path sampling and other enhanced sampling techniques allows us to capture the rare event of water exchange in the first hydration shell of Mg$^{2+}$ revealing a dissociative exchange mechanism. Transition interface sampling yields exchange rates within one order of magnitude of experiment, representing a substantial improvement over classical dissociative force fields. In contrast, the NNP-derived solvation free energy significantly underestimates the experimental value, revealing a limitation of the present local NNP architectures for describing ion solvation thermodynamics. Our results demonstrate that DFT-trained NNPs can accurately describe Mg$^{2+}$ hydration structure, diffusion, ion pairing, and exchange kinetics, while highlighting the need for explicit long-range electrostatic treatments to achieve quantitative agreement with experimental ion solvation free energies.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops MACE neural network potentials trained on revPBE-D3/zd and revPBE0-D3/zd DFT data for aqueous MgCl2 solutions. It benchmarks these NNPs on experimental properties including first-shell structure, diffusion coefficients, ion pairing, water-exchange rates and mechanism (via transition path sampling and transition interface sampling), and solvation free energy. The central claims are that the NNPs accurately reproduce structure, diffusion, pairing, and exchange kinetics (rates within one order of magnitude of experiment, dissociative mechanism), while underestimating solvation free energy due to limitations of local architectures in treating long-range electrostatics.
Significance. If the results hold after validation, the work would demonstrate that DFT-trained local NNPs can substantially improve upon classical force fields for kinetic properties such as rare water-exchange events in Mg2+ while exposing a clear limitation for thermodynamic quantities. The combination of NNPs with TPS/TIS for enhanced sampling of exchange paths is a methodological strength that enables direct comparison to experimental rates.
major comments (2)
- [Abstract] Abstract: the claim that transition interface sampling yields exchange rates within one order of magnitude of experiment rests on the untested assumption that the MACE NNP remains accurate for the high-energy, partially dissociated Mg–water geometries that define the dissociative transition state. Standard equilibrium AIMD reference data (revPBE-D3/zd and revPBE0-D3/zd) rarely sample these configurations; local message-passing architectures can produce uncontrolled extrapolation errors outside the training distribution. This is load-bearing for the kinetics result and requires explicit validation (e.g., NNP vs. DFT energies/forces on TPS-generated TS-like snapshots).
- [Abstract] Abstract, final paragraph: the reported significant underestimation of the experimental solvation free energy is presented without quantitative comparison values, error bars, or details on the free-energy calculation protocol. This weakens the attribution to “local NNP architectures” and prevents assessment of whether the discrepancy is systematic or within the expected uncertainty of the method.
minor comments (1)
- [Abstract] Abstract: no training-set size, validation RMSE, or error bars on any reported quantities (structure, diffusion, rates) are provided, which reduces the ability to judge the precision of the claimed agreements.
Simulated Author's Rebuttal
Thank you for your thorough review and insightful comments. We address the major comments point-by-point below, agreeing where revisions are warranted.
read point-by-point responses
-
Referee: Abstract: the claim that transition interface sampling yields exchange rates within one order of magnitude of experiment rests on the untested assumption that the MACE NNP remains accurate for the high-energy, partially dissociated Mg–water geometries that define the dissociative transition state. Standard equilibrium AIMD reference data (revPBE-D3/zd and revPBE0-D3/zd) rarely sample these configurations; local message-passing architectures can produce uncontrolled extrapolation errors outside the training distribution. This is load-bearing for the kinetics result and requires explicit validation (e.g., NNP vs. DFT energies/forces on TPS-generated TS-like snapshots).
Authors: We agree that explicit validation on TS-like configurations is necessary to support the kinetics claims, as equilibrium AIMD training data may not fully cover these regions. In the revised manuscript we will add a direct NNP-vs-DFT comparison of energies and forces on TPS-generated snapshots near the dissociative transition state to quantify any extrapolation errors. revision: yes
-
Referee: Abstract, final paragraph: the reported significant underestimation of the experimental solvation free energy is presented without quantitative comparison values, error bars, or details on the free-energy calculation protocol. This weakens the attribution to “local NNP architectures” and prevents assessment of whether the discrepancy is systematic or within the expected uncertainty of the method.
Authors: We agree that additional quantitative details are required. The revised manuscript will include the specific solvation free energy values with error bars, a full description of the free-energy protocol, and an expanded discussion of the experimental comparison to clarify whether the discrepancy is systematic or within expected uncertainties. revision: yes
Circularity Check
No circularity: predictions validated against independent external data
full rationale
The paper trains MACE NNPs on external DFT reference calculations (revPBE-D3/zd and revPBE0-D3/zd) and directly compares the resulting models to independent experimental measurements for hydration structure, diffusion coefficients, ion pairing, water-exchange rates (obtained via transition path sampling and transition interface sampling), and solvation free energies. No step reduces a claimed prediction to a fitted parameter or self-citation by construction; the derivation chain remains self-contained against external benchmarks with no self-definitional, fitted-input, or load-bearing self-citation patterns present.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption revPBE-D3/zd and revPBE0-D3/zd DFT calculations supply accurate reference data for training NNPs on aqueous MgCl2
- domain assumption Transition path sampling combined with the NNP can correctly identify the dissociative water-exchange mechanism and rate
Reference graph
Works this paper leans on
-
[1]
V. K. Misra and D. E. Draper, On the role of magnesium ions in RNA stability, Biopolymers48, 113 (1998)
1998
-
[2]
N. H. Williams, Magnesium Ion Catalyzed ATP Hydroly- sis, Journal of the American Chemical Society122, 12023 (2000)
2000
-
[3]
Cowan, Structural and catalytic chemistry of magnesium-dependent enzymes, Biometals15, 225 (2002)
J. Cowan, Structural and catalytic chemistry of magnesium-dependent enzymes, Biometals15, 225 (2002)
2002
-
[4]
Pyle, Metal ions in the structure and function of RNA, JBIC Journal of Biological Inorganic Chemistry7, 679 (2002)
A. Pyle, Metal ions in the structure and function of RNA, JBIC Journal of Biological Inorganic Chemistry7, 679 (2002). 10
2002
-
[5]
B. Born, H. Weing¨ artner, E. Br¨ undermann, and M. Havenith, Solvation Dynamics of Model Peptides Probed by Terahertz Spectroscopy. Observation of the Onset of Collective Network Motions, Journal of the American Chemical Society131, 3752 (2009)
2009
-
[6]
Stachura, S
M. Stachura, S. Chakraborty, A. Gottberg, L. Ruck- thong, V. L. Pecoraro, and L. Hemmingsen, Direct Ob- servation of Nanosecond Water Exchange Dynamics at a Protein Metal Site, Journal of the American Chemical Society139, 79 (2017)
2017
-
[7]
Alln´ er, L
O. Alln´ er, L. Nilsson, and A. Villa, Magnesium Ion–Water Coordination and Exchange in Biomolecular Simulations, Journal of Chemical Theory and Computa- tion8, 1493 (2012)
2012
-
[8]
Li and K
P. Li and K. M. Merz, Taking into Account the Ion- Induced Dipole Interaction in the Nonbonded Model of Ions, Journal of Chemical Theory and Computation10, 289 (2014)
2014
-
[9]
Dubou´ e-Dijon, P
E. Dubou´ e-Dijon, P. E. Mason, H. E. Fischer, and P. Jungwirth, Hydration and Ion Pairing in Aqueous Mg 2+ and Zn 2+ Solutions: Force-Field Description Aided by Neutron Scattering Experiments and Ab Initio Molecular Dynamics Simulations, The Journal of Physi- cal Chemistry B122, 3296 (2018)
2018
-
[10]
Mamatkulov and N
S. Mamatkulov and N. Schwierz, Force fields for mono- valent and divalent metal cations in TIP3P water based on thermodynamic and kinetic properties, The Journal of Chemical Physics148, 074504 (2018)
2018
-
[11]
K. K. Grotz, S. Cruz-Le´ on, and N. Schwierz, Opti- mized Magnesium Force Field Parameters for Biomolec- ular Simulations with Accurate Solvation, Ion-Binding, and Water-Exchange Properties, Journal of Chemical Theory and Computation17, 2530 (2021)
2021
-
[12]
Soniat, L
M. Soniat, L. Hartman, and S. W. Rick, Charge Trans- fer Models of Zinc and Magnesium in Water, Journal of Chemical Theory and Computation11, 1658 (2015)
2015
-
[13]
Z. Jing, C. Liu, R. Qi, and P. Ren, Many-body effect determines the selectivity for Ca 2+ and Mg 2+ in pro- teins, Proceedings of the National Academy of Sciences 115, 10.1073/pnas.1805049115 (2018)
-
[14]
S. Mamatkulov, M. Fyta, and R. R. Netz, Force fields for divalent cations based on single-ion and ion- pair properties, The Journal of Chemical Physics138, 10.1063/1.4772808 (2013)
-
[15]
P. Li, B. P. Roberts, D. K. Chakravorty, and K. M. Merz, Rational Design of Particle Mesh Ewald Compat- ible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent, Journal of Chemical Theory and Com- putation9, 2733 (2013)
2013
-
[16]
Schwierz, Kinetic pathways of water exchange in the first hydration shell of magnesium, The Journal of Chem- ical Physics152, 224106 (2020)
N. Schwierz, Kinetic pathways of water exchange in the first hydration shell of magnesium, The Journal of Chem- ical Physics152, 224106 (2020)
2020
-
[17]
Cruz-Le´ on and N
S. Cruz-Le´ on and N. Schwierz, Hofmeister Series for Metal-Cation–RNA Interactions: The Interplay of Bind- ing Affinity and Exchange Kinetics, Langmuir36, 5979 (2020)
2020
-
[18]
M. Fyta and R. R. Netz, Ionic force field optimization based on single-ion and ion-pair solvation properties: Go- ing beyond standard mixing rules, The Journal of Chem- ical Physics136, 10.1063/1.3693330 (2012)
-
[19]
Leontyev and A
I. Leontyev and A. Stuchebrukhov, Accounting for elec- tronic polarization in non-polarizable force fields, Physi- cal Chemistry Chemical Physics13, 2613 (2011)
2011
-
[20]
Kohagen, P
M. Kohagen, P. E. Mason, and P. Jungwirth, Accurate Description of Calcium Solvation in Concentrated Aque- ous Solutions, The Journal of Physical Chemistry B118, 7902 (2014)
2014
-
[21]
I. M. Zeron, J. L. F. Abascal, and C. Vega, A force field of Li+, Na+, K+, Mg2+, Ca2+, Cl-, and SO42- in aqueous solution based on the TIP4P/2005 water model and scaled charges for the ions, The Journal of Chemical Physics151, 10.1063/1.5121392 (2019)
-
[22]
P. Li, L. F. Song, and K. M. Merz, Systematic Parame- terization of Monovalent Ions Employing the Nonbonded Model, Journal of Chemical Theory and Computation 11, 1645 (2015)
2015
-
[23]
C. Schran, F. L. Thiemann, P. Rowe, E. A. M¨ uller, O. Marsalek, and A. Michaelides, Machine learning po- tentials for complex aqueous systems made simple, Pro- ceedings of the National Academy of Sciences118, 10.1073/pnas.2110077118 (2021)
-
[24]
Kocer, T
E. Kocer, T. W. Ko, and J. Behler, Neural Network Po- tentials: A Concise Overview of Methods, Annual Review of Physical Chemistry73, 163 (2022)
2022
-
[25]
A. Omranpour, P. Montero de Hijes, J. Behler, and C. Dellago, Perspective: Atomistic simulations of water and aqueous systems with machine learning potentials, The Journal of Chemical Physics160, 10.1063/5.0201241 (2024)
-
[26]
J. Daru, H. Forbert, J. Behler, and D. Marx, Coupled Cluster Molecular Dynamics of Condensed Phase Sys- tems Enabled by Machine Learning Potentials: Liquid Water Benchmark, Physical Review Letters129, 226001 (2022)
2022
-
[27]
J. Liu, J. Lan, and X. He, Toward High-level Machine Learning Potential for Water Based on Quantum Frag- mentation and Neural Networks, The Journal of Physical Chemistry A126, 3926 (2022)
2022
-
[28]
Batatia, D
I. Batatia, D. P. Kov´ acs, G. N. Simm, C. Ortner, and G. Cs´ anyi, MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields, inAdvances in Neural Information Processing Systems, Vol. 35 (2022)
2022
-
[29]
Morawietz, A
T. Morawietz, A. Singraber, C. Dellago, and J. Behler, How van der Waals interactions determine the unique properties of water, Proceedings of the National Academy of Sciences113, 8368 (2016)
2016
-
[30]
P. Montero de Hijes, C. Dellago, R. Jinnouchi, B. Schmiedmayer, and G. Kresse, Comparing machine learning potentials for water: Kernel-based regression and Behler–Parrinello neural networks, The Journal of Chemical Physics160, 10.1063/5.0197105 (2024)
-
[31]
F. C. Lightstone, E. Schwegler, R. Q. Hood, F. Gygi, and G. Galli, A first principles molecular dynamics simulation of the hydrated magnesium ion, Chemical Physics Letters 343, 549 (2001)
2001
-
[32]
Bhattacharjee, A
A. Bhattacharjee, A. B. Pribil, B. R. Randolf, B. M. Rode, and T. S. Hofer, Hydration of Mg2+ and its influ- ence on the water hydrogen bonding network via ab initio QMCF MD, Chemical Physics Letters536, 39 (2012)
2012
-
[33]
X. Wang, D. Toroz, S. Kim, S. L. Clegg, G.-S. Park, and D. D. Tommaso, Density functional theory based molecular dynamics study of solution composition effects on the solvation shell of metal ions, Physical Chemistry Chemical Physics22, 16301 (2020)
2020
-
[34]
Juraskova, G
V. Juraskova, G. Tusha, H. Zhang, L. V. Sch¨ afer, and F. Duarte, Modelling ligand exchange in metal complexes 11 with machine learning potentials, Faraday Discussions 256, 156 (2025)
2025
-
[35]
Ferretti, G
A. Ferretti, G. Melani, L. Benedetti, R. A. Sorodoc, A. Fortunelli, and G. Brancato, Accurate Simulations of Water and Aqueous Solutions through Fine-Tuned Dispersion-Corrected Density Functional Theory and Machine-Learning Interatomic Potentials, Journal of Chemical Information and Modeling65, 12437 (2025)
2025
-
[36]
O’Neill, B
N. O’Neill, B. X. Shi, K. Fong, A. Michaelides, and C. Schran, To Pair or not to Pair? Machine-Learned Explicitly-Correlated Electronic Structure for NaCl in Water, The Journal of Physical Chemistry Letters15, 6081 (2024)
2024
-
[37]
Soyemi and T
A. Soyemi and T. Szilv´ asi, Modeling the Behavior of Complex Aqueous Electrolytes Using Machine Learning Interatomic Potentials: The Case of Sodium Sulfate, The Journal of Physical Chemistry B129, 9405 (2025)
2025
-
[38]
C. Cao, A. Kingan, R. C. Hill, J. Kuang, L. Wang, C. Zhang, M. R. Carbone, H. van Dam, S. Yoo, S. Yan, E. S. Takeuchi, K. J. Takeuchi, X. Wu, A. M. Abeykoon, A. C. Marschilok, and D. Lu, Resolving the Solvation Structure and Transport Properties of Aqueous Zinc Electrolytes from Salt-in-Water to Water-in-Salt Using Neural Network Potential, PRX Energy4, 0...
2025
-
[39]
M. J. Gillan, D. Alf` e, and A. Michaelides, Perspective: How good is DFT for water?, The Journal of Chemical Physics144, 10.1063/1.4944633 (2016)
-
[40]
Palos, E
E. Palos, E. Lambros, S. Swee, J. Hu, S. Dasgupta, and F. Paesani, Assessing the Interplay between Functional- Driven and Density-Driven Errors in DFT Models of Wa- ter, Journal of Chemical Theory and Computation18, 3410 (2022)
2022
-
[41]
Dasgupta, E
S. Dasgupta, E. Lambros, J. P. Perdew, and F. Pae- sani, Elevating density functional theory to chemical ac- curacy for water simulations through a density-corrected many-body formalism, Nature Communications12, 6359 (2021)
2021
-
[42]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Physical Review Letters77, 3865 (1996)
1996
-
[43]
Generalized Gra- dient Approximation Made Simple
Y. Zhang and W. Yang, Comment on “Generalized Gra- dient Approximation Made Simple”, Physical Review Letters80, 890 (1998)
1998
-
[44]
Adamo and V
C. Adamo and V. Barone, Toward reliable density func- tional methods without adjustable parameters: The PBE0 model, The Journal of Chemical Physics110, 6158 (1999)
1999
-
[45]
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A con- sistent and accurate ab initio parametrization of den- sity functional dispersion correction (DFT-D) for the 94 elements H-Pu, The Journal of Chemical Physics132, 10.1063/1.3382344 (2010)
-
[46]
Grimme, S
S. Grimme, S. Ehrlich, and L. Goerigk, Effect of the damping function in dispersion corrected density func- tional theory, Journal of Computational Chemistry32, 1456 (2011)
2011
-
[47]
K. N. Lausch, R. E. Haouari, D. Trzewik, and J. Behler, Impact of the damping function in dispersion-corrected density functional theory on the properties of liq- uid water, The Journal of Chemical Physics163, 10.1063/5.0275244 (2025)
-
[48]
M. J. Abraham, T. Murtola, R. Schulz, S. P´ all, J. C. Smith, B. Hess, and E. Lindahl, GROMACS: High per- formance molecular simulations through multi-level par- allelism from laptops to supercomputers, SoftwareX1-2, 19 (2015)
2015
-
[49]
W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein, Comparison of simple poten- tial functions for simulating liquid water, The Journal of Chemical Physics79, 926 (1983)
1983
-
[50]
Hutter, M
J. Hutter, M. Iannuzzi, F. Schiffmann, and J. VandeVon- dele, cp2k: atomistic simulations of condensed matter systems, WIREs Computational Molecular Science4, 15 (2014)
2014
-
[51]
Lippert, J
G. Lippert, J. Hutter, and M. Parrinello, The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations, Theoretical Chemistry Accounts: Theory, Computation, and Mod- eling (Theoretica Chimica Acta)103, 124 (1999)
1999
-
[52]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, Separable dual- space Gaussian pseudopotentials, Physical Review B54, 1703 (1996)
1996
-
[53]
Hartwigsen, S
C. Hartwigsen, S. Goedecker, and J. Hutter, Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Physical Review B58, 3641 (1998)
1998
-
[55]
P. Montero de Hijes, C. Dellago, R. Jinnouchi, and G. Kresse, Density isobar of water and melting tem- perature of ice: Assessing common density functionals, The Journal of Chemical Physics161, 10.1063/5.0227514 (2024)
-
[56]
Eastman, J
P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Sim- monett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks, and V. S. Pande, OpenMM 7: Rapid de- velopment of high performance algorithms for molecular dynamics, PLOS Computational Biology13, e1005659 (2017)
2017
-
[57]
D. A. Sivak, J. D. Chodera, and G. E. Crooks, Time Step Rescaling Recovers Continuous-Time Dynamical Proper- ties for Discrete-Time Langevin Integration of Nonequi- librium Systems, The Journal of Physical Chemistry B 118, 6466 (2014)
2014
-
[58]
Laio and M
A. Laio and M. Parrinello, Escaping free-energy min- ima, Proceedings of the National Academy of Sciences 99, 12562 (2002)
2002
-
[59]
Barducci, G
A. Barducci, G. Bussi, and M. Parrinello, Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method, Physical Review Letters100, 020603 (2008)
2008
-
[60]
Torrie and J
G. Torrie and J. Valleau, Nonphysical sampling distri- butions in Monte Carlo free-energy estimation: Um- brella sampling, Journal of Computational Physics23, 187 (1977)
1977
-
[61]
Bonomi, G
M. Bonomi, G. Bussi, C. Camilloni, G. A. Tribello, P. Ban´ aˇ s, A. Barducci, M. Bernetti, P. G. Bolhuis, S. Bot- taro, D. Branduardi, R. Capelli, P. Carloni, M. Ceriotti, A. Cesari, H. Chen, W. Chen, F. Colizzi, S. De, M. D. L. Pierre, D. Donadio, V. Drobot, B. Ensing, A. L. Fer- guson, M. Filizola, J. S. Fraser, H. Fu, P. Gasparotto, F. L. Gervasio, F. ...
2019
-
[62]
G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni, and G. Bussi, PLUMED 2: New feathers for an old bird, Computer Physics Communications185, 604 (2014)
2014
-
[63]
J. G. Kirkwood and F. P. Buff, The Statistical Mechan- ical Theory of Solutions. I, The Journal of Chemical Physics19, 774 (1951)
1951
-
[64]
Karwounopoulos, Z
J. Karwounopoulos, Z. Wu, S. Tkaczyk, S. Wang, A. Baskerville, K. Ranasinghe, T. Langer, G. P. F. Wood, M. Wieder, and S. Boresch, Insights and Challenges in Correcting Force Field Based Solvation Free Energies Us- ing a Neural Network Potential, The Journal of Physical Chemistry B128, 6693 (2024)
2024
-
[65]
A. K. Picha, S. Tkaczyk, T. Langer, M. Wieder, and S. Boresch, Architecture-Independent Absolute Solvation Free Energy Calculations with Neural Network Poten- tials, The Journal of Physical Chemistry Letters16, 12080 (2025)
2025
-
[66]
P. G. Bolhuis, D. Chandler, C. Dellago, and P. L. Geissler, Transition Path Sampling: Throwing Ropes Over Rough Mountain Passes, in the Dark, Annual Re- view of Physical Chemistry53, 291 (2002)
2002
-
[67]
T. S. van Erp, D. Moroni, and P. G. Bolhuis, A novel path sampling method for the calculation of rate constants, The Journal of Chemical Physics118, 7762 (2003)
2003
-
[68]
T. S. van Erp, Reaction Rate Calculation by Paral- lel Path Swapping, Physical Review Letters98, 268301 (2007)
2007
-
[69]
Kumar, J
S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen, and P. A. Kollman, THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method, Journal of Computational Chemistry13, 1011 (1992)
1992
-
[70]
Marcus, Ionic radii in aqueous solutions, Chemical Reviews88, 1475 (1988)
Y. Marcus, Ionic radii in aqueous solutions, Chemical Reviews88, 1475 (1988)
1988
-
[71]
Marcus,Ion Properties(Marcel Dekker, 1997)
Y. Marcus,Ion Properties(Marcel Dekker, 1997)
1997
-
[72]
R. A. Robinson and R. H. Stokes,Electrolyte Solutions, 2nd ed. (Dover Publications, 2002)
2002
-
[73]
Bleuzen, P.-A
A. Bleuzen, P.-A. Pittet, L. Helm, and A. E. Merbach, Water exchange on magnesium(II) in aqueous solution: a variable temperature and pressure17O NMR study, Mag- netic Resonance in Chemistry35, 765 (1997)
1997
-
[74]
Neely and R
J. Neely and R. Connick, Rate of water exchange from hydrated magnesium ion, Journal of the American Chem- ical Society92, 3476 (1970)
1970
-
[75]
Helm and A
L. Helm and A. Merbach, Water exchange on metal ions: experiments and simulations, Coordination Chemistry Reviews187, 151 (1999)
1999
-
[76]
Mondal, D
A. Mondal, D. Kussainova, S. Yue, and A. Z. Pana- giotopoulos, Modeling Chemical Reactions in Alkali Car- bonate–Hydroxide Electrolytes with Deep Learning Po- tentials, Journal of Chemical Theory and Computation 19, 4584 (2023)
2023
- [77]
-
[78]
D. Jiao, C. King, A. Grossfield, T. A. Darden, and P. Ren, Simulation of Ca 2+ and Mg 2+ Solvation Using Polarizable Atomic Multipole Potential, The Journal of Physical Chemistry B110, 18553 (2006)
2006
-
[79]
Yeh and G
I.-C. Yeh and G. Hummer, System-Size Dependence of Diffusion Coefficients and Viscosities from Molecular Dy- namics Simulations with Periodic Boundary Conditions, The Journal of Physical Chemistry B108, 15873 (2004)
2004
-
[80]
Falkner and N
S. Falkner and N. Schwierz, Kinetic pathways of water exchange in the first hydration shell of magnesium: In- fluence of water model and ionic force field, The Journal of Chemical Physics155, 084503 (2021)
2021
-
[81]
H. B. G. C. H. Langford,Ligand Substitution Processes (W. A. Benjamin, Inc., 1965)
1965
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.