High-Throughput Computation of Anharmonic Low-Frequency Protein Vibrations
Pith reviewed 2026-05-19 09:42 UTC · model grok-4.3
The pith
Coarse-graining all-atom protein trajectories lets FRESEAN analysis extract anharmonic low-frequency vibrations at far lower cost.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Coarse-graining of all-atom simulation trajectories can be combined with FRESEAN mode analysis to extract information on low-frequency vibrations at minimal computational cost.
What carries the argument
Coarse-graining of all-atom molecular dynamics trajectories before applying FRESEAN mode analysis, where FRESEAN isolates anharmonic low-frequency modes from velocity time correlation functions without harmonic approximations.
If this is right
- Low-frequency vibrations become accessible for larger proteins than full all-atom analysis previously allowed.
- The resulting modes retain their value as collective variables for enhanced sampling of conformational ensembles.
- High-throughput studies of anharmonic vibrations in folded proteins become practical.
- Applications to future conformational transition work are directly supported.
Where Pith is reading between the lines
- The same coarse-graining step could be tested on other large biomolecules such as nucleic acids or membrane complexes.
- Comparing the coarse-grained modes against experimental far-infrared spectra on the same proteins would provide an external check.
- If the modes continue to work as collective variables, the method could support routine screening of how sequence changes affect slow protein motions.
Load-bearing premise
Coarse-graining the trajectories keeps the essential low-frequency anharmonic vibrational signals intact and free of major artifacts.
What would settle it
Direct comparison on a small protein showing that the low-frequency modes from the coarse-grained trajectory differ substantially in frequency or direction from those of the original all-atom trajectory.
Figures






read the original abstract
At room temperature, low frequency vibrations at far-infrared frequencies are thermally excited ($k_B T > h \nu$) and not restricted to harmonic fluctuations around a single potential energy minimum. For folded proteins, these intrinsically anharmonic vibrations can contain information on slow conformational transitions. Recently, we have developed FREquency-SElective ANharmonic (FRESEAN) mode analysis, a method based on time correlation functions that isolates low-frequency vibrational motions from molecular dynamics simulation trajectories without relying on harmonic approximations. We recently showed that low-frequency vibrations obtained from FRESEAN mode analysis are effective collective variables in enhanced sampling simulations of conformational ensembles. However, FRESEAN mode analysis is based on velocity time correlations between all degrees of freedom, which creates computational challenges for large biomolecules. To facilitate future applications, we demonstrate here how coarse-graining of all-atom simulation trajectories can be combined with FRESEAN mode analysis to extract information on low-frequency vibrations at minimal computational cost.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes combining coarse-graining of all-atom molecular dynamics trajectories with FRESEAN mode analysis to extract anharmonic low-frequency vibrational modes in proteins at reduced computational cost, extending prior FRESEAN work for applications to larger biomolecules where full all-atom velocity correlations are prohibitive.
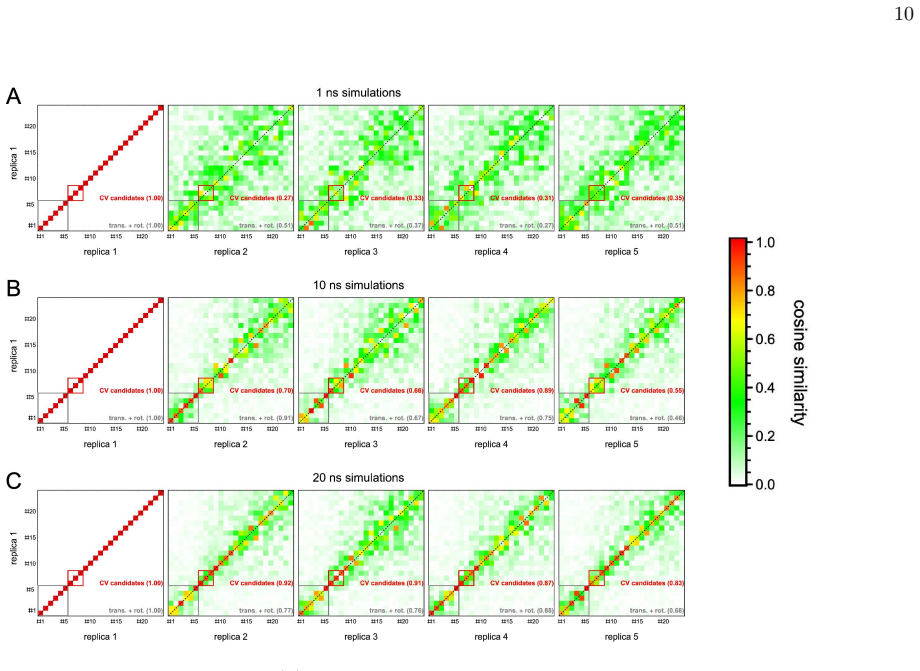
Significance. If the coarse-graining step is shown to retain the essential non-Gaussian velocity statistics and cross-correlations that encode anharmonicity, the approach would enable scalable computation of collective variables for enhanced sampling of conformational ensembles in large proteins, directly addressing the scaling limitation noted in the abstract.
major comments (2)
- [Abstract / Methods] Abstract and § Methods (coarse-graining description): The central efficiency claim rests on the untested assertion that spatial averaging to Cα or residue centers preserves the velocity time-correlation functions used by FRESEAN; no quantitative overlap metrics (e.g., mode similarity scores or anharmonicity diagnostics) between all-atom and coarse-grained FRESEAN results on identical trajectories are reported, leaving the preservation step load-bearing but unsupported.
- [Results] Results section (demonstration examples): The reported cost reduction is shown only for a small number of systems; without error bars on mode frequencies or direct comparison of the extracted low-frequency spectra to all-atom FRESEAN on the same data, it is unclear whether the coarse-grained modes remain faithful for the intended use as collective variables in enhanced sampling.
minor comments (2)
- [Methods] Notation for the coarse-graining operator should be defined explicitly (e.g., as a projection matrix) rather than described only in prose.
- [Figures] Figure captions should state the protein systems, simulation lengths, and coarse-graining level used in each panel.
Simulated Author's Rebuttal
We thank the referee for the constructive comments. We respond to each major point below and describe the revisions that will be made to address the concerns raised.
read point-by-point responses
-
Referee: [Abstract / Methods] Abstract and § Methods (coarse-graining description): The central efficiency claim rests on the untested assertion that spatial averaging to Cα or residue centers preserves the velocity time-correlation functions used by FRESEAN; no quantitative overlap metrics (e.g., mode similarity scores or anharmonicity diagnostics) between all-atom and coarse-grained FRESEAN results on identical trajectories are reported, leaving the preservation step load-bearing but unsupported.
Authors: We agree that the current manuscript does not include quantitative metrics comparing all-atom and coarse-grained velocity correlations or mode overlaps. In the revised manuscript we will add a new subsection (or supplementary figure) that reports direct comparisons on at least one test trajectory, including mode-vector overlaps and differences in the extracted low-frequency spectra. This addition will provide the requested evidence that the essential non-Gaussian statistics are retained after coarse-graining. revision: yes
-
Referee: [Results] Results section (demonstration examples): The reported cost reduction is shown only for a small number of systems; without error bars on mode frequencies or direct comparison of the extracted low-frequency spectra to all-atom FRESEAN on the same data, it is unclear whether the coarse-grained modes remain faithful for the intended use as collective variables in enhanced sampling.
Authors: The referee is correct that the present results lack error estimates and side-by-side spectral comparisons. We will revise the Results section to include (i) error bars derived from multiple independent trajectories and (ii) a direct overlay or table of low-frequency spectra obtained from both all-atom and coarse-grained FRESEAN on the same data sets. These additions will clarify the fidelity of the coarse-grained modes for subsequent use as collective variables. revision: yes
Circularity Check
Minor self-citation to prior FRESEAN work; combination claim independent of self-definition or fitted inputs.
full rationale
The paper's derivation chain consists of referencing the authors' prior FRESEAN development (self-citation) and then proposing its combination with standard coarse-graining of trajectories to reduce computational cost for low-frequency mode extraction. This self-citation is not load-bearing for the central efficiency claim, which rests on the methodological combination rather than any equation that reduces a 'prediction' to a fitted parameter or redefines the output in terms of the input. No self-definitional steps, ansatz smuggling, or renaming of known results appear in the provided text; the preservation of anharmonic content under coarse-graining is stated as an assumption for future validation, not derived circularly. The work remains self-contained against external benchmarks such as direct all-atom comparisons.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Molecular dynamics trajectories provide representative sampling of anharmonic low-frequency vibrations in folded proteins.
- ad hoc to paper Coarse-graining retains sufficient information about low-frequency modes for FRESEAN analysis.
Reference graph
Works this paper leans on
-
[1]
provides a novel perspective on low-frequency vi- brations in molecules, which does not rely on harmonic approximations. Instead of mapping all degrees of free- dom to a single set of orthogonal vibrational coordinates, dynamic fluctuations in a simulation trajectory are ana- lyzed as a function of frequency. As a result, orthogonal sets of collective vib...
work page 1960
-
[2]
Y. Miao, V. A. Feher, and J. A. McCammon, Gaus- sian accelerated molecular dynamics: Unconstrained en- hanced sampling and free energy calculation, J. Chem. Theory Comput. 11, 3584 (2015)
work page 2015
-
[3]
Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett. 314, 141 (1999)
work page 1999
-
[4]
G. Torrie and J. Valleau, Nonphysical sampling distri- butions in monte carlo free-energy estimation: Umbrella sampling, J. Comput. Phys. 23, 187 (1977)
work page 1977
-
[5]
A. Laio and M. Parrinello, Escaping free-energy minima, Proc. Natl. Acad. Sci. U.S.A 99, 12562 (2002)
work page 2002
-
[6]
D. M. Zuckerman and L. T. Chong, Weighted ensem- ble simulation: review of methodology, applications, and software, Annu. Rev. Biophys. 46, 43 (2017)
work page 2017
-
[7]
C. C. David and D. J. Jacobs, Principal component anal- ysis: a method for determining the essential dynamics of proteins, Methods Mol Biol. 1084, 193 (2014)
work page 2014
-
[8]
Y. Naritomi and S. Fuchigami, Slow dynamics of a pro- tein backbone in molecular dynamics simulation revealed by time-structure based independent component analy- sis, J. Chem. Phys. 139, 215102 (2013)
work page 2013
-
[9]
S. Schultze and H. Grubm¨ uller, Time-lagged independent component analysis of random walks and protein dynam- 7 ics, J. Chem. Theory Comput. 17, 5766 (2021), pMID: 34449229
work page 2021
-
[10]
Z. Zou, D. Wang, and P. Tiwary, A graph neural network- state predictive information bottleneck (gnn-spib) ap- proach for learning molecular thermodynamics and ki- netics, Digit. Discov. 4, 211 (2025)
work page 2025
-
[11]
A. Majumder and J. E. Straub, Machine learning derived collective variables for the study of protein homodimer- ization in membrane, J. Chem. Theory Comput.20, 5774 (2024), pMID: 38918177
work page 2024
-
[12]
D. Mendels, G. Piccini, and M. Parrinello, Collective variables from local fluctuations, J. Phys. Chem. Lett. 9, 2776 (2018), pMID: 29733652
work page 2018
-
[13]
H. Chen, H. Liu, H. Feng, H. Fu, W. Cai, X. Shao, and C. Chipot, Mlcv: Bridging machine-learning-based dimensionality reduction and free-energy calculation, J. Chem. Inf. Model. 62, 1 (2022), pMID: 34939790
work page 2022
-
[14]
S. Chatterjee and D. Ray, Acceleration with inter- pretability: A surrogate model-based collective variable for enhanced sampling, J. Chem. Theory Comput. 21, 1561 (2025)
work page 2025
-
[15]
C. Chennubhotla, A. J. Rader, L.-W. Yang, and I. Bahar, Elastic network models for understanding biomolecular machinery: from enzymes to supramolecular assemblies, Phys. Biol. 2, S173 (2005)
work page 2005
-
[16]
M. M. Tirion, Large amplitude elastic motions in pro- teins from a single-parameter, atomic analysis, Phys. Rev. Lett. 77, 1905 (1996)
work page 1905
- [17]
-
[18]
T. Haliloglu, I. Bahar, and B. Erman, Gaussian dynamics of folded proteins, Phys. Rev. Lett. 79, 3090 (1997)
work page 1997
-
[19]
B. Brooks and M. Karplus, Harmonic dynamics of pro- teins: normal modes and fluctuations in bovine pancre- atic trypsin inhibitor, Proc. Natl. Acad. Sci. U.S.A 80, 6571 (1983)
work page 1983
-
[20]
S. Mahajan and Y.-H. Sanejouand, On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins, Arch. Biochem. Bio- phys. 567, 59 (2015)
work page 2015
-
[21]
T. D. Romo, A. Grossfield, and A. G. Markelz, Persistent protein motions in a rugged energy landscape revealed by normal mode ensemble analysis, J. Chem. Inf. Model.60, 6419 (2020)
work page 2020
-
[22]
R. Levy, A. Srinivasan, W. Olson, and J. McCammon, Quasi-harmonic method for studying very low frequency modes in proteins, Biopolymers 23, 1099 (1984)
work page 1984
-
[23]
R. M. Levy, O. De la Luz Rojas, and R. A. Friesner, Quasi-harmonic method for calculating vibrational spec- tra from classical simulations on multi-dimensional an- harmonic potential surfaces, J. Phys. Chem. 88, 4233 (1984)
work page 1984
-
[24]
M. A. Sauer, S. Mondal, B. Neff, S. Maiti, and M. Hey- den, Fast sampling of protein conformational dynamics, (2024), arXiv:2411.08154
work page internal anchor Pith review Pith/arXiv arXiv 2024
- [25]
-
[27]
M. J. Abraham, T. Murtola, R. Schulz, S. P´ all, J. C. Smith, B. Hess, and E. Lindahl, Gromacs: High perfor- mance molecular simulations through multi-level paral- lelism from laptops to supercomputers, SoftwareX 1, 19 (2015)
work page 2015
- [28]
-
[29]
M. G. S. Costa, P. R. Batista, P. M. Bisch, and D. Per- ahia, Exploring free energy landscapes of large conforma- tional changes: molecular dynamics with excited normal modes, J. Chem. Theory Comput. 11, 2755 (2015)
work page 2015
-
[30]
K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror, and D. E. Shaw, Improved side- chain torsion potentials for the amber ff99sb protein force field, Proteins: Struct. Funct. Bioinf. 78, 1950 (2010)
work page 1950
-
[31]
W.-L. Jorgensen, J. Chandrasekhar, J.-D. Madura, R.- W. Impey, and M.-L. Klein, Comparison of simple po- tential functions for simulating liquid water, J. Chem. Phys. 79, 926 (1983)
work page 1983
- [32]
-
[33]
M. Bernetti and G. Bussi, Pressure control using stochas- tic cell rescaling, J. Chem. Phys. 153, 10.1063/5.0020514 (2020)
-
[34]
Nos´ e, A molecular dynamics method for simulations in the canonical ensemble, Mol
S. Nos´ e, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys. 52, 255 (1984)
work page 1984
-
[35]
W. G. Hoover, Canonical dynamics: Equilibrium phase- space distributions, Phys. Rev. A 31, 1695 (1985)
work page 1985
-
[36]
M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys. 52, 7182 (1981)
work page 1981
-
[37]
B. Hess, H. Bekker, H. J. Berendsen, and J. G. Fraaije, Lincs: A linear constraint solver for molecular simula- tions, J. Comput. Chem. 18, 1463 (1997)
work page 1997
- [38]
-
[39]
M. A. Sauer and M. Heyden, Frequency-selective anhar- monic mode analysis of thermally excited vibrations in proteins, J. Chem. Theory Comput. 19, 5481 (2023)
work page 2023
-
[40]
S. Chakraborty, S.-K. Sinha, and S. Bandyopadhyay, Low-frequency vibrational spectrum of water in the hy- dration layer of a protein: A molecular dynamics simu- lation study, J. Phys. Chem. B 111, 13626 (2007)
work page 2007
- [41]
-
[42]
G. Mathias and M. D. Baer, Generalized normal coordi- nates for the vibrational analysis of molecular dynamics simulations, J. Chem. Theory Comput. 7, 2028 (2011)
work page 2028
-
[43]
M. A. Sauer, S. Mondal, and M. Heyden, FRESEAN- metadynamics DOI: 10.5281/zenodo.15678841 (2025)
-
[44]
M. Heyden, FRESEAN-tutorial DOI: 10.5281/zen- odo.15686127 (2025)
-
[45]
A. Lerbret, A. H´ edoux, B. Annigh¨ ofer, and M.-C. Bellissent-Funel, Influence of pressure on the low- frequency vibrational modes of lysozyme and water: A complementary inelastic neutron scattering and molecu- lar dynamics simulation study, Proteins: Struct. Funct. Bioinf. 81, 326 (2012)
work page 2012
-
[46]
M. Heyden and D. J. Tobias, Spatial dependence of protein-water collective hydrogen-bond dynamics, Phys. 8 Rev. Lett. 111, 218101 (2013)
work page 2013
-
[47]
Zwanzig, Time-correlation functions and transport coefficients in statistical mechanics, Annu
R. Zwanzig, Time-correlation functions and transport coefficients in statistical mechanics, Annu. Rev. Phys. Chem. 16, 67 (1965)
work page 1965
-
[48]
A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dun- brack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, et al. , All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B 102, 3586 (1998)
work page 1998
-
[49]
M. R. Krebs, D. K. Wilkins, E. W. Chung, M. C. Pitkeathly, A. K. Chamberlain, J. Zurdo, C. V. Robin- son, and C. M. Dobson, Formation and seeding of amy- loid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain, J. Mol. Biol. 300, 541 (2000)
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.