GPU-accelerated Modeling of Biological Regulatory Networks
Pith reviewed 2026-05-19 09:46 UTC · model grok-4.3
The pith
GPU implementations of global optimization algorithms accelerate parameter searches for biological logic models by 33 to 1866 percent over CPU methods.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
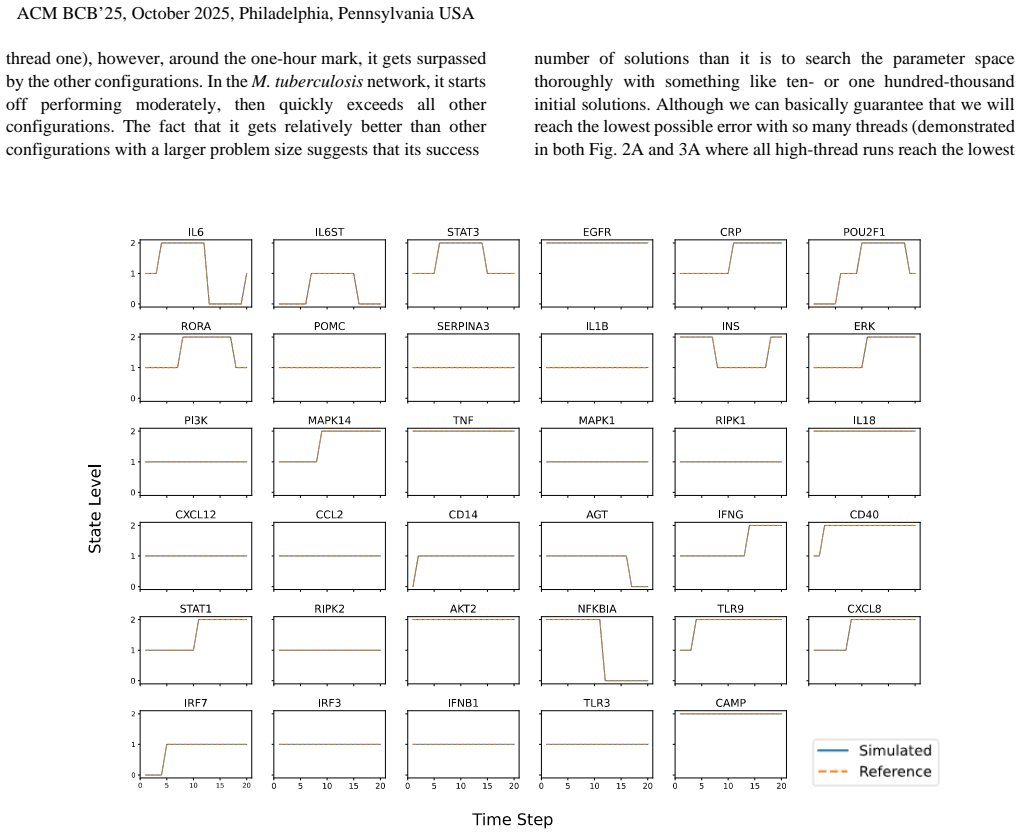
The implementation of global optimization algorithms in a GPU-computing environment accelerates the solution of parameter search problems for discrete logic models of biological regulatory networks, yielding 33%-43% improvement compared to multi-thread CPU implementations and 33%-1866% increase compared to CPU in serial on two model systems that represent almost an order of magnitude scale-up in complexity.
What carries the argument
GPU-accelerated global optimization applied to fitting discrete logic models to time-course data from regulatory networks
If this is right
- Parameter searches become feasible for regulatory networks of greater size without requiring prohibitive runtimes.
- Researchers can generate predictions about network behavior under varying conditions on shorter timescales.
- In silico hypothesis generation and experiment design gain viability as routine tools in pharmaceutical research.
- Model identification from sparse data can support iterative refinement of regulatory hypotheses within practical time limits.
Where Pith is reading between the lines
- The same GPU porting strategy could be tested on continuous or hybrid models of regulatory dynamics to check for comparable gains.
- Smaller labs without large CPU clusters might now run ensemble fits that quantify uncertainty in the recovered logic rules.
- Integration with automated experiment design loops could become realistic if the reduced runtimes allow many candidate models to be evaluated per cycle.
Load-bearing premise
The two chosen model biological regulatory systems are representative of the scale and structure of networks that would be used in practical in silico pharmaceutical research so the observed speedups generalize without major algorithm changes.
What would settle it
Running identical parameter searches on a third regulatory network at least twice as large as the larger of the two tested models and checking whether the GPU-to-CPU runtime ratios remain inside the reported 33-1866 percent improvement band.
Figures




read the original abstract
The complex regulatory dynamics of a biological network can be succinctly captured using discrete logic models. Given even sparse time-course data from the system of interest, previous work has shown that global optimization schemes are suitable for proposing logic models that explain the data and make predictions about how the system will behave under varying conditions. Considering the large scale of the parameter search spaces associated with these regulatory systems, performance optimizations on the level of both hardware and software are necessary for making this a practical tool for in silico pharmaceutical research. We show here how the implementation of these global optimization algorithms in a GPU-computing environment can accelerate the solution of these parameter search problems considerably. We carry out parameter searches on two model biological regulatory systems that represent almost an order of magnitude scale-up in complexity, and we find the gains in efficiency from GPU to be a 33%-43% improvement compared to multi-thread CPU implementations and a 33%-1866% increase compared to CPU in serial. These improvements make global optimization of logic model identification a far more attractive and feasible method for in silico hypothesis generation and design of experiments.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that GPU implementations of global optimization algorithms for inferring discrete logic models of biological regulatory networks from time-course data yield substantial speedups, specifically 33%-43% over multi-threaded CPU baselines and 33%-1866% over serial CPU on two model systems representing nearly an order-of-magnitude increase in complexity; these gains are presented as making the approach practical for in silico pharmaceutical research.
Significance. If the reported speedups are substantiated with reproducible benchmarks, the work would be significant for systems biology by addressing the computational bottleneck of large parameter spaces in logic-based network modeling, potentially enabling wider adoption for hypothesis generation and experiment design. The emphasis on hardware-level optimizations for sparse regulatory systems is a timely contribution, though the absence of detailed empirical support limits immediate impact assessment.
major comments (2)
- [Abstract] Abstract: the concrete performance claims (33%-43% improvement over multi-thread CPU and up to 1866% over serial) are stated without any benchmark tables, error bars, implementation details, or description of how the multi-thread CPU baseline was configured (e.g., thread count, hardware specs, or optimization flags). This directly undermines support for the central empirical claim of considerable acceleration.
- [Results] The manuscript tests only two model systems and does not examine whether the observed GPU speedups generalize to larger node counts, denser interaction graphs, or more complex logic functions typical of pharmaceutical-scale networks; memory divergence or host-device transfer overheads could alter efficiency, but no scaling analysis or stress tests on such regimes are provided.
minor comments (2)
- [Methods] Notation for the logic functions and parameter search space could be clarified with an explicit example in the methods to aid readers unfamiliar with discrete regulatory models.
- [Abstract] The abstract mentions 'almost an order of magnitude scale-up in complexity' but does not quantify this (e.g., number of nodes, edges, or parameters) in a table or equation for easy comparison.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and constructive comments on our manuscript. We have addressed each major comment point by point below, providing clarifications and indicating revisions where the manuscript will be updated to strengthen the presentation of our results.
read point-by-point responses
-
Referee: [Abstract] Abstract: the concrete performance claims (33%-43% improvement over multi-thread CPU and up to 1866% over serial) are stated without any benchmark tables, error bars, implementation details, or description of how the multi-thread CPU baseline was configured (e.g., thread count, hardware specs, or optimization flags). This directly undermines support for the central empirical claim of considerable acceleration.
Authors: We agree that the abstract would benefit from additional context to support the performance claims. The detailed benchmark tables, including timings with error bars, hardware specifications (NVIDIA GPU model and multi-core CPU with explicit thread counts), and optimization flags (e.g., -O3 for CPU and CUDA compilation settings), are presented in the Results section along with the experimental setup. To address this, we have revised the abstract to include a brief reference to the hardware configurations used for the multi-threaded CPU baseline and to direct readers to the full benchmark details and tables in the main text. revision: yes
-
Referee: [Results] The manuscript tests only two model systems and does not examine whether the observed GPU speedups generalize to larger node counts, denser interaction graphs, or more complex logic functions typical of pharmaceutical-scale networks; memory divergence or host-device transfer overheads could alter efficiency, but no scaling analysis or stress tests on such regimes are provided.
Authors: The two model systems were selected specifically because they represent nearly an order-of-magnitude increase in complexity over prior examples, providing a practical demonstration of the GPU approach on representative biological regulatory networks. We acknowledge that the current study does not include a comprehensive scaling analysis across larger node counts, denser graphs, or more complex logic functions, nor explicit stress tests for memory divergence and host-device transfer overheads. We have added a new paragraph in the Discussion section that explicitly addresses these potential limitations, discusses the relevance of such factors for pharmaceutical-scale applications, and outlines directions for future work on generalization and performance characterization in those regimes. revision: partial
Circularity Check
Empirical runtime benchmarks contain no circular derivation
full rationale
The paper reports measured wall-clock speedups (33-43% over multi-thread CPU, up to 1866% over serial) obtained by executing the same global optimization code on GPU versus CPU hardware for two concrete logic-model identification tasks. These figures are direct experimental observations, not quantities derived from equations, fitted parameters, or prior self-citations that would reduce to the inputs by construction. No self-definitional loops, uniqueness theorems, or ansatz smuggling appear in the derivation chain; the central performance claims remain externally falsifiable by re-running the reported benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Global optimization schemes are suitable for proposing logic models that explain the data and make predictions
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We show here how the implementation of these global optimization algorithms in a GPU-computing environment can accelerate the solution of these parameter search problems considerably... simulated annealing algorithm... objective function y = min[( |S(x) − R| ) ⊙ ( |G| + 1) ]
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanLogicNat unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
discrete logic formulation... state values of nodes can take on one of three activation values... priority update scheme
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Yuan Mei, Maya L. Gosztyla, Xinzhu Tan, et al. 2025. Integrated multi-omic characterizations of the synapse reveal RNA processing factors and ubiquitin ligases associated with neurodevelopmental disorders. Cell Syst 16, 4, Article 101204 (April 2025). http://dx.doi.org/10.1016/j.cels.2025.101204
-
[2]
Serrano, Adrian Pelin, Tabiwang N
Lia R. Serrano, Adrian Pelin, Tabiwang N. Arrey, et al. 2025. Affinity purification mass spectrometry on the Orbitrap-Astral mass spectrometer enables ACM BCB’25, October 2025, Philadelphia, Pennsylvania USA J. Reimer et al. high-throughput protein-protein interaction mapping. J. Proteome Res 24, 4 (March 2025), 2006-2016. https://doi.org/10.1021/acs.jpro...
-
[3]
Alexiou Athanasios, Vairaktarakis Charalampos, Tsiamis Vasileios and Ghulam Md Ashraf. 2017. Protein-protein interaction (PPI) network: recent advances in drug discovery. Curr. Drug Metab 18, 1 (Jan. 2017), 5-10. https://doi.org/10.2174/138920021801170119204832
-
[4]
Craddock, Darrell Whitley and Gordon Broderick
Hooman Sedghamiz, Matthew Morris, Travis J.A. Craddock, Darrell Whitley and Gordon Broderick. 2019. Bio-ModelChecker: using bounded constraint satisfaction to seamlessly integrate observed behavior with prior knowledge of biological networks. Front. Bioeng. Biotechnol 7, 48 (March 2019). https://doi.org/10.3389/fbioe.2019.00048
-
[5]
René Thomas and Richard d’Ari. 1990. Biological Feedback. CRC Press, Inc
work page 1990
-
[6]
Monteiro, Julio Saez- Rodriguez, Tomá š Helikar, Denis Thieffry and Claudine Chaouiya
Wassim Abou-Jaoudé, Pauline Traynard, Pedro T. Monteiro, Julio Saez- Rodriguez, Tomá š Helikar, Denis Thieffry and Claudine Chaouiya. 2016. Logical modeling and dynamical analysis of cellular networks. Front. Genet 7, 94 (May 2016). https://doi.org/10.3389/fgene.2016.00094
-
[7]
Jonas Béal, Lorenzo Pantolini, Vincent Noël, Emmanuel Barillot and Laurence Calzone. 2021. Personalized logical models to investigate cancer response to BRAF treatments in melanomas and colorectal cancers. Comput. Biol 17, 1, Article e1007900 (Jan. 2021). https://doi.org/10.1371/journal.pcbi.1007900
-
[8]
Nicola Bonzanni, Abhishek Garg, K. Anton Feenstra et al. 2013. Hard-wired heterogeneity in blood stem cells revealed using a dynamic regulatory network model. Bioinformatics 29, 13 (July 2013), i80-888. https://doi.org/10.1093/bioinformatics/btt243
-
[9]
Cole A. Lyman, Matthew M. Morris, Spencer Richman, Hongbao Cao, Antony Scerri, Chris Cheadle and Gordon Broderick. 2021. High fidelity modeling of pulse dynamics using logic networks. In 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), December 9-12, 2021, Houston, Texas. IEEE, 197-204. https://doi.org/10.1109/BIBM52615.2021.9669437
-
[10]
Anton Dekkers and Emile Aarts. 1991. Global optimization and simulated annealing. Math. Progr 50 (March 1991), 367-393. https://doi.org/10.1007/BF01594945
-
[11]
S. Kirkpatrick, C.D. Gelatt, Jr. and M.P. Vecchi. 1983. Optimization by simulated annealing. Science 220, 4598 (May 1983), 671-680. https://doi.org/10.1126/science.220.4598.671
-
[12]
A.M. Ferreiro, J.A. García, J.G. López-Salas and C. Vásquez. 2012. An efficient implementation of parallel simulated annealing algorithm in GPUs. J. Glob. Optim, 57 (Sept. 2012), 863-890. https://doi.org/10.1007/s10898-012-9979-z
-
[13]
Tias Guns. 2019. Increasing modeling language convenience with a universal n- dimensional array, CPpy as python-embedded example. In Proceedings of the 18th workshop on Constraint Modelling and Reformulation at CP (ModRef 2019), September 30, 2019, Stamford, Connecticut
work page 2019
-
[14]
Siu Kwan Lam, Antoine Pitrou and Stanley Seibert. 2015. Numba: a llvm-based python jit compiler. In Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC (LLVM ’15), November 15, 2015, Austin, Texas. ACM Inc., New York, NY, 1-6. https://doi.org/10.1145/2833157.2833162
-
[15]
Robert I. McLachlan, Nancy L. Cohen, Kristine D. Dahl, William J. Bremner and Michael R. Soules. 1990. Serum inhibin levels during the periovulatory interval in normal women: relationships with sex steroid and gonadotrophin levels. Clin. Endocrinol (Oxf) , 32, 1 (Jan. 1990), 39-48. https://doi.org/10.1111/j.1365- 2265.1990.tb03748.x
-
[16]
Bappaditya Dey and William R. Bishai. 2014. Crosstalk between Mycobacterium tuberculosis and the host cell. Semin. Immunol, 26, 6 (Dec. 2014), 486-496. https://doi.org/10.1016/j.smim.2014.09.002
-
[17]
John A. Bachman, Benjamin M. Gyori and Peter K. Sorger. 2023. Automated assembly of molecular mechanisms at scale from text mining and curated databases. Mol Syst. Biol , 19, Article e11325 (March 2023). https://doi.org/10.15252/msb.202211325
-
[18]
Jiuhai Chen and Jonas Mueller. 2024. Quantifying uncertainty in answers from any language model and enhancing their trustworthiness. In Proceedings of the 62nd Annual Meeting of the Association for Computational Linguistics (Volume 1:Long Papers) , August 2024, Bangkok, Thailand. Association for Computational Linguistics, 5186-5200. https://doi.org/10.186...
-
[19]
Tair Askar, Bekdaulet Shukirgaliyev, Martin Lukac and Ernazar Abdikamalov
-
[20]
Computation, 9, 12, Article 142 (December 2021)
Evaluation of pseudo-random number generation on GPU cards. Computation, 9, 12, Article 142 (December 2021). https://doi.org/10.3390/computation9120142
-
[21]
M. Manssen, M. Weigel and A.K. Hartmann. 2012. Random number generators for massively parallel simulations on GPU. Eur. Phys. J. Spec. Top, 210 (September 2012), 53-71. https://doi.org/10.1140/epjst/e2012-01637-8
-
[22]
Emran Bajrami, Maida Ašić, Emir Cogo, Dino Trnka and Novica Nosovic. 2012. Performance comparison of simulated annealing algorithm execution on GPU and CPU. In 2012 Proceedings of the 35th International Convention MIPRO , May 21-25, 2012, Opatija, Croatia. IEEE, 1785-1788
work page 2012
-
[23]
Davis, Erik Verschueren, Gwendolyn M
Zoe H. Davis, Erik Verschueren, Gwendolyn M. Jang, et al. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell , 57, 2 (Jan. 2015), 349-60. https://doi.org/10.1016/j.molcel.2014.11.026
-
[24]
Bennett H. Penn, Zoe Netter, Jeffrey R. Johnson et al. 2018. An Mtb-human protein-protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol. Cell, 71, 4 (Aug. 2018), 637-648. https://doi.org/10.1016/j.molcel.2018.07.010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.