Electron-molecule scattering via R-matrix variational algorithms on a quantum computer
Pith reviewed 2026-05-19 05:20 UTC · model grok-4.3
The pith
Variational quantum algorithms solve the inner-region R-matrix problem for electron-molecule scattering and encode the needed boundary amplitudes in circuit parameters.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
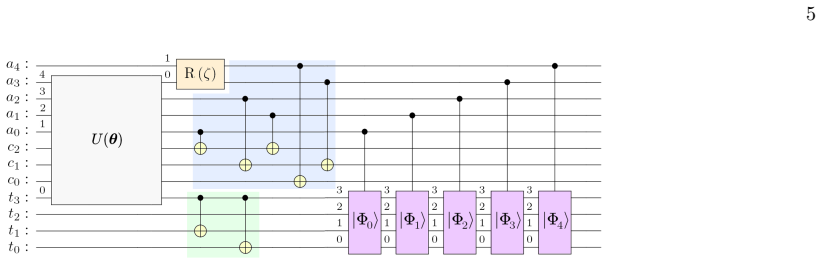
The R-matrix inner-region problem is formulated on a quantum computer by using the variational quantum eigensolver (and variants with number projection and simultaneous optimisation) to diagonalise the inner-region Hamiltonian. For electron scattering from the hydrogen molecule in a selected symmetry sector, the procedure recovers the complete spectrum and shows that the optimal circuit parameters directly encode the boundary amplitudes needed for the outer-region matching and cross-section computation.
What carries the argument
The variational quantum eigensolver applied to the inner-region Hamiltonian, in which the optimised parameters of the circuit ansatz directly supply the R-matrix boundary amplitudes.
If this is right
- The full spectrum recovered inside the chosen symmetry sector provides all states required for a complete scattering description.
- The encoded boundary amplitudes can be fed directly into standard outer-region propagation and matching routines.
- Number-projection operators can be added to enforce physical constraints such as fixed electron number.
- Simultaneous optimisation of circuit parameters can accelerate convergence to the required boundary values.
Where Pith is reading between the lines
- The same variational formulation could be applied to other diatomic or small polyatomic targets once suitable ansatze are identified.
- Running the algorithm on present-day noisy quantum processors would reveal the practical error tolerance for scattering observables.
- If hardware noise remains manageable, the method could open scattering studies of molecules whose inner-region basis sizes exceed classical diagonalisation limits.
Load-bearing premise
That variational quantum eigensolver results obtained on a noiseless simulator for a small basis set will produce boundary amplitudes accurate enough for the outer-region scattering calculation to remain reliable on noisy hardware or for larger molecules.
What would settle it
A direct numerical comparison, for the same hydrogen-molecule model and basis, between the boundary amplitudes extracted from the optimal VQE circuit parameters and those obtained from a conventional classical R-matrix calculation; a mismatch larger than the target precision would falsify the encoding claim.
Figures




read the original abstract
Electron-molecule collisions play a central role in both natural processes and modern technological applications, particularly in plasma processing. Conventional computational strategies such as the R-matrix method have been widely adopted yet encounter significant scaling challenges in treating more complex systems. In this work we present a quantum computational approach that utilises the variational quantum eigensolver (VQE) and variations thereof to overcome these limitations. We explore a number of methods, including the use of number projection operators and simultaneous optimisation. We demonstrate the feasibility of our method on a model problem of electron scattering from the hydrogen molecule, with numerical results obtained using a noiseless classical simulator. We recover the full spectrum of the Hamiltonian within a chosen symmetry sector. Moreover, the optimal circuit parameters directly encode the R-matrix boundary amplitudes needed for subsequent scattering computations. To our knowledge, this is the first application of quantum algorithms to electron--molecule scattering, and specifically the first formulation of the R-matrix inner-region problem on a quantum computer.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes applying the variational quantum eigensolver (VQE), including variants with number projection and simultaneous optimization, to solve the inner-region Hamiltonian in the R-matrix method for electron-molecule scattering. For the model problem of electron-H2 scattering, results on a noiseless classical simulator recover the full spectrum within a chosen symmetry sector. The authors assert that the optimal circuit parameters directly encode the R-matrix boundary amplitudes needed for outer-region scattering calculations, presenting this as the first quantum-algorithm treatment of electron-molecule scattering.
Significance. If the extracted boundary amplitudes prove accurate and the approach scales beyond small noiseless cases, it could alleviate the basis-size and configuration-interaction scaling bottlenecks that limit classical R-matrix calculations for larger molecules. The spectrum recovery on the H2 model supplies concrete evidence of feasibility for the inner-region step in a limited setting and credits the work as an initial demonstration of quantum methods in this domain.
major comments (2)
- Numerical results: spectrum recovery is demonstrated, yet no comparison is provided between the boundary amplitudes read out from the optimal VQE parameters and the corresponding amplitudes obtained from classical diagonalization of the same inner-region Hamiltonian. Without this check, agreement on eigenvalues does not establish that the surface amplitudes are sufficiently accurate for reliable R-matrix construction and subsequent scattering observables.
- Method and results sections: the manuscript contains no noisy-hardware simulations or error-propagation analysis for the extracted amplitudes. This omission leaves untested the weakest assumption that noiseless-simulator amplitudes will remain usable when the same ansatz is executed on actual quantum hardware or for larger molecules.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments. We address each major point below and describe the revisions that will be made to strengthen the manuscript.
read point-by-point responses
-
Referee: Numerical results: spectrum recovery is demonstrated, yet no comparison is provided between the boundary amplitudes read out from the optimal VQE parameters and the corresponding amplitudes obtained from classical diagonalization of the same inner-region Hamiltonian. Without this check, agreement on eigenvalues does not establish that the surface amplitudes are sufficiently accurate for reliable R-matrix construction and subsequent scattering observables.
Authors: We agree that explicit validation of the boundary amplitudes is required to substantiate the claim that the optimal parameters encode them. Although spectrum recovery indicates that the variational states approximate the eigenstates, this does not automatically confirm the surface amplitudes. In the revised manuscript we will extract the R-matrix boundary amplitudes from the converged VQE parameters and provide a direct, quantitative comparison to the amplitudes obtained from classical diagonalization of the identical inner-region Hamiltonian, including overlap or relative-error metrics. revision: yes
-
Referee: Method and results sections: the manuscript contains no noisy-hardware simulations or error-propagation analysis for the extracted amplitudes. This omission leaves untested the weakest assumption that noiseless-simulator amplitudes will remain usable when the same ansatz is executed on actual quantum hardware or for larger molecules.
Authors: The present work is a proof-of-principle demonstration performed on a noiseless simulator. We acknowledge that noise resilience must be examined for practical use. In the revision we will add an error-propagation analysis that quantifies how parameter uncertainties propagate to the extracted boundary amplitudes. Full noisy-hardware simulations and scaling tests for larger molecules lie outside the scope of this initial study and will be addressed in follow-up work. revision: partial
Circularity Check
No circularity: standard VQE readout of boundary amplitudes from inner-region optimization
full rationale
The paper formulates the R-matrix inner-region problem as a standard VQE on the Hamiltonian for a chosen symmetry sector of electron-H2, recovers the spectrum on a noiseless simulator, and extracts boundary amplitudes directly from the optimized circuit parameters. This extraction follows the usual variational state readout and does not reduce to a self-definition, fitted input renamed as prediction, or self-citation chain; the amplitudes are a direct consequence of the variational wavefunction rather than being presupposed by the scattering result. No uniqueness theorems, ansatzes smuggled via prior work, or renamings of known patterns are load-bearing. The derivation remains self-contained against the classical R-matrix inner-region benchmark.
Axiom & Free-Parameter Ledger
free parameters (1)
- ansatz circuit depth and form
axioms (2)
- domain assumption The R-matrix method partitions space into an inner region where exchange and correlation are treated explicitly and an outer region where the scattered electron moves in a long-range potential
- domain assumption The variational quantum eigensolver can recover the low-lying eigenstates of the inner-region Hamiltonian when run on a noiseless simulator
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the optimal circuit parameters directly encode the R-matrix boundary amplitudes needed for subsequent scattering computations
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We recover the full spectrum of the Hamiltonian within a chosen symmetry sector
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Present a quantum-algorithm framework for the R- matrix inner-region problem, mapping the ( N + 1)-electron Hamiltonian to qubits via a Jor- dan–Wigner transformation and explicitly enforc- ing occupancy constraints for continuum orbitals
-
[2]
Employ the qubit representation of the number- projection operator in the variational cir- cuit—based on the construction in [27]—to ensure that at most one electron occupies any continuum orbital
-
[3]
Develop a sequential subspace-optimisation proto- col that decomposes the required SO( N) rotations into a cascade of two-qubit Givens rotations, allow- arXiv:2507.05514v1 [quant-ph] 7 Jul 2025 2 ing us to converge on each eigenstate with signif- icantly fewer cost-function evaluations and Pauli- term measurements than alternative methods
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[4]
Introduce a coherent-summation measurement scheme for the target-eigenstate register, which combines ancilla-based Hadamard gates with system-Hamiltonian measurements to reduce the total number of distinct expectation values re- quired
-
[5]
Demonstrate our approach on a model H 2 scatter- ing calculation, validating the algorithm and show- ing that the optimal circuit parameters directly en- code the R-matrix boundary amplitudes needed for subsequent scattering analysis. II. THE R-MA TRIX METHOD AND MOLECULAR HAMIL TONIAN The R-matrix method was originally developed to study nuclear [28], an...
-
[6]
A parametric rotation gate U(θ), followed by the Clebsch–Gordan rotations R(ζ) that are fixed by 4 spin symmetry (in our example, as in Figure 1, a single rotation corresponding to the triplet state). R(ζ) is a fixed two-qubit gate (implemented as a Givens rotation of angle ζ) that couples target eigenstate qubits to enforce total-spin symmetry
-
[7]
The target–continuum couplings (blue block in Fig- ure 1, CNOT gates with control on the target- eigenstate register and target on the continuum reg- ister). Each target eigenstate qubit acts as control on one continuum orbital qubit to ensure that, if a particular target eigenfunction is selected, exactly one continuum orbital is excited in the correct s...
-
[8]
The bound-states gate (green block in Figure 1, CNOT gates with control and targets on the target- orbital register). A gate that entangles the target eigenstate qubits with the target-orbital qubits, so that if the circuit has selected a particular bound configuration, the correct pattern of target-orbital occupations is realised. In simple examples, one...
-
[9]
The sequence of controlled target-eigenstate gates, each controlled on one of the target eigenstate qubits and with targets on the target-orbital qubits. A central element of our quantum circuits is the one- parameter two-qubit rotation operation which mixes the |01⟩ and the |10⟩ states, implemented through a sequence of controlled-NOT (CNOT) gates interl...
-
[10]
Whilst the coupling between the upper multiplet with St = S − 1 2 and the lower multiplet with St = S + 1 2 is dynamical, the couplings between states with Mt = M − 1 2 and Mt = M + 1 2 for total spin in the upper multiplet and the lower multiplet are each fixed by symmetry. Each spin multiplet admits a two-term expansion in terms of the half-angle ζ: S, ...
-
[11]
(15) Where H 2 has been replaced by HP H to enforce correct occupancy
Single state with variance cost function In the single eigenstate approach the cost function is defined as the variance of the Hamiltonian with respect to the trial state, C(θ) = ⟨HP H ⟩|ψ(θ)⟩ − ⟨H⟩2 |ψ(θ)⟩. (15) Where H 2 has been replaced by HP H to enforce correct occupancy. This sometimes fails to return the full eigenspectrum
-
[12]
Single state with folded Hamiltonian cost function An alternative approach is to first evaluate the en- ergy expectation for each trial state: the approxi- mate energy ˜E is first evaluated for the trial state, and then used to construct a Hamiltonian [43, 44] where, as long as the objective state eigenvalue is the closest eigenvalue to the energy of the ...
-
[13]
Simultaneous optimisation with sum of vari- ances In the simultaneous optimisation of all five eigen- states (see explicit list in Results section), the cost function is constructed as the sum of the variances of the individual eigenstates, C(θ) = 5X i=1 h ⟨HP H ⟩|ψi(θ)⟩ − ⟨H⟩2 |ψi(θ)⟩ i . (17) This approach, while necessitating separate evalua- tions for...
-
[14]
Simultaneous optimisation with subspace optimisation and simple Hamiltonian expec- tation value The most computational resource efficient method is based solely on the Hamiltonian expectation value, Ci(θi,i+1, . . . , θi,N −1) = ⟨H⟩|ψi(θ)⟩. (18) In this approach the unitary is designed to enforce a subspace structure that naturally preserves or- thogonali...
-
[15]
Single state with variance cost function In some instances this approach is unable to recover all the eigenvalues
-
[16]
The results are shown in the upper panel of Figure 3
Single state with folded Hamiltonian cost function This approach required approximately 1,397 cost function evaluations to recover the five eigenstates, each corresponding to the evaluation of 1,080 Pauli terms. The results are shown in the upper panel of Figure 3
-
[17]
The results are shown in the middle panel of Figure 3
Simultaneous optimisation with sum of vari- ances This method required 13,345 cost function evalua- tions for 1,080 Pauli terms. The results are shown in the middle panel of Figure 3
-
[18]
The results are shown in lower panel of Figure 3
Simultaneous optimisation with subspace optimisation and simple Hamiltonian expec- tation value This method required only 573 cost function eval- uations involving 92 Pauli terms. The results are shown in lower panel of Figure 3. For the last three algorithms all the eigenvalues were recovered as the converged values within 10 −7Ha, the residual discrepan...
-
[19]
B. I. Schneider, T. N. Rescigno, B. H. Lengsfield, and A. E. Orel, Science 284, 1489 (1999)
work page 1999
-
[20]
J. M. Carr, P. G. Galiatsatos, J. D. Gorfinkiel, M. A. Lysaght, Z. Masin, L. A. Morgan, I. Rozum, and J. Ten- nyson, Eur. Phys. J. D 66, 58 (2012)
work page 2012
-
[21]
M. C. Zammit, J. S. Savage, D. V. Fursa, and I. Bray, Phys. Rev. Lett. 116, 233201 (2016)
work page 2016
-
[22]
K. Bartschat and M. J. Kushner, Proc. Nat. Acad. Sci. 113, 7026 (2016)
work page 2016
-
[23]
R. P. Feynman, Int. J. Theor. Phys. 21, 467 (1982)
work page 1982
- [24]
-
[25]
D. S. Abrams and S. Lloyd, Phys. Rev. Lett. 79, 2586 (1997)
work page 1997
-
[26]
D. S. Abrams and S. Lloyd, Phys. Rev. Lett. 83, 5162 (1999). 9
work page 1999
-
[27]
A. Aspuru-Guzik, A. D. Dutoi, P. J. Love, and M. Head- Gordon, Science 309, 1704 (2005)
work page 2005
-
[28]
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’Brien, Nat. Commun. 5, 4213 (2014)
work page 2014
-
[29]
J. R. McClean, J. Romero, R. Babbush, and A. Aspuru- Guzik, New J. Phys. 18, 023023 (2016)
work page 2016
-
[30]
B. P. Lanyon, J. D. Whitfield, G. G. Gillett, M. E. Goggin, M. P. Almeida, I. Kassal, J. D. Biamonte, M. Mohseni, B. J. Powell, M. Barbieri, A. Aspuru-Guzik, and A. G. White, Nat. Chem. 2, 106 (2010)
work page 2010
-
[31]
P. J. J. O’Malley, R. Babbush, I. D. Kivlichan, J. Romero, J. R. McClean, R. Barends, J. Kelly, P. Roushan, A. Tranter, N. Ding, B. Campbell, Y. Chen, Z. Chen, B. Chiaro, A. Dunsworth, A. G. Fowler, E. Jef- frey, A. Megrant, J. Y. Mutus, C. Neill, C. Quintana, D. Sank, A. Vainsencher, J. Wenner, T. C. White, P. V. Coveney, P. J. Love, H. Neven, A. Aspuru-...
work page 2016
- [32]
-
[33]
Y. Cao, J. Romero, J. P. Olson, M. Degroote, P. D. John- son, M. Kieferov´ a, I. D. Kivlichan, T. Menke, B. Per- opadre, N. P. D. Sawaya, S. Sim, L. Veis, and A. Aspuru- Guzik, Chem. Rev. 119, 10856 (2019)
work page 2019
-
[34]
S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin, and X. Yuan, Rev. Mod. Phys. 92, 015003 (2020)
work page 2020
- [35]
- [36]
-
[37]
K. Bharti, A. Cervera-Lierta, T. H. Kyaw, T. Haug, S. Alperin-Lea, A. Anand, M. Degroote, H. Heimonen, K. Fujii, A. Y. Matsuura, M. Singh, P. Murali, K. Lauter, T. Brandenburger, D. E. Koh, O. Shehab, S. Wood, A. Siripurapu, F. Nori, N. Killoran, H. Neven, A. Aspuru- Guzik, S. Pandey, A. Jardine, A. Greene, J. McClean, Z. Wang, I. Tavernelli, C. Lin, and ...
work page 2022
-
[38]
S. P. Jordan, K. S. M. Lee, and J. Preskill, arXiv preprint arXiv:1112.4833 (2011)
work page internal anchor Pith review Pith/arXiv arXiv 2011
-
[39]
S. P. Jordan, K. S. M. Lee, and J. Preskill, Science 336, 1130 (2012)
work page 2012
-
[40]
S. P. Jordan, K. S. M. Lee, and J. Preskill, arXiv preprint arXiv:1404.7115 (2014)
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[41]
C. Choi, D. Lee, J. Bonitati, Z. Qian, and J. Watkins, Phys. Rev. Lett. 127, 040505 (2021)
work page 2021
- [42]
- [43]
-
[44]
M. Yusf, L. Gan, C. Moffat, and G. Ru- pak, arXiv preprint arXiv:2406.09231 (2024), 10.48550/arXiv.2406.09231, arXiv:2406.09231 [nucl- th]
-
[45]
Pascal’s pyramid and number projec- tion operators for quantum computation,
D. Picozzi, “Pascal’s pyramid and number projec- tion operators for quantum computation,” (2024), arXiv:2407.16561 [quant-ph]
-
[46]
E. P. Wigner and L. Eisenbud, Phys. Rev. 72, 29 (1947)
work page 1947
-
[47]
P. G. Burke, R-Matrix Theory of Atomic Collisions: Ap- plication to Atomic, Molecular and Optical Processes (Springer, 2011)
work page 2011
- [48]
-
[49]
Z. Maˇ s´ ın, J. Benda, J. D. Gorfinkiel, A. G. Harvey, and J. Tennyson, Comput. Phys. Commun. 249, 107092 (2020)
work page 2020
-
[50]
A. G. Sunderland, J. W. Heggarty, C. J. Noble, and N. S. Scott, Comput. Phys. Commun. 114, 183 (1998)
work page 1998
-
[51]
J. D. Gorfinkiel and J. Tennyson, J. Phys. B 37, L343 (2004)
work page 2004
-
[52]
J. D. Gorfinkiel and J. Tennyson, J. Phys. B 38, 1607 (2005)
work page 2005
- [53]
- [54]
- [55]
- [56]
-
[57]
J. D. Whitfield, J. Biamonte, and A. Aspuru-Guzik, Mol. Phys. 109, 735 (2011)
work page 2011
- [58]
- [59]
-
[60]
D. W. Berry, C. Gidney, M. Motta, J. R. McClean, and R. Babbush, npj Quantum Inf. 4, 22 (2018)
work page 2018
- [61]
-
[62]
L. C. Tazi and A. J. W. Thom, Journal of Chemical The- ory and Computation 20, 2491 (2024)
work page 2024
-
[63]
M. J. D. Powell, in Advances in Optimization and Nu- merical Analysis, Mathematics and Its Applications, Vol. 275, edited by S. Gomez and J.-P. Hennart (Kluwer Aca- demic Publishers, Dordrecht, 1994) pp. 51–67
work page 1994
-
[64]
M. J. D. Powell, Acta Numerica 7, 287 (1998)
work page 1998
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.