Inference of maximum parsimony phylogenetic trees with model-based classical and quantum methods
Pith reviewed 2026-05-19 01:40 UTC · model grok-4.3
The pith
Three optimization models let classical and quantum solvers search all tree topologies and ancestral states for maximum parsimony phylogenetic trees.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
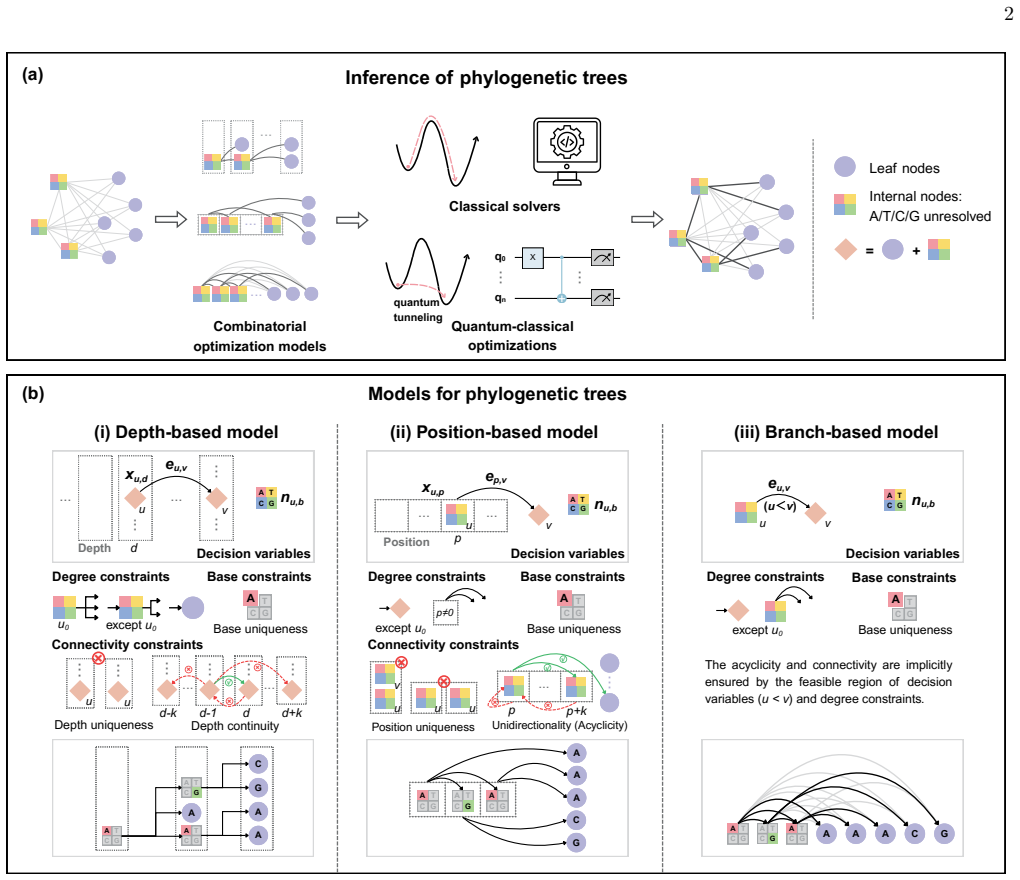
The authors design three optimization models compatible with classical and quantum solvers that directly search the complete solution space of all possible tree topologies and ancestral states. Among them the branch-based model drastically reduces the number of variables and explicit constraints through a specific variable definition. The model is validated by a classical solver that obtains solutions generally better than heuristics on the GAPDH gene dataset, while quantum simulations find the exact optimal solutions for small-scale instances with rapid convergence.
What carries the argument
The branch-based model, which encodes tree structures and ancestral states with a variable definition that drastically reduces the number of variables and explicit constraints.
If this is right
- The branch-based modeling approach applies directly to other tree optimization problems beyond phylogenetics.
- Quantum solvers can recover exact optimal maximum-parsimony trees for small instances.
- Direct enumeration of topologies and states removes biases that arise when candidate internal nodes are pre-constructed.
- Classical implementations of the model can produce lower-parsimony trees than common heuristic methods on real gene datasets.
Where Pith is reading between the lines
- Larger quantum processors could extend the approach to phylogenetic instances too big for exhaustive classical search.
- The variable-reduction technique may transfer to related combinatorial tree problems in bioinformatics and network design.
- Testing the models on additional datasets with known ground-truth trees would clarify whether the classical gains generalize.
Load-bearing premise
The optimization models correctly encode the maximum parsimony criterion by directly enumerating all tree topologies and ancestral states without introducing new biases, and the classical solver results on the GAPDH dataset reflect true improvements over heuristics.
What would settle it
Running the classical solver on the GAPDH gene dataset and comparing the obtained parsimony scores against published heuristic benchmarks; if the scores are not lower, the claim of generally better solutions does not hold.
Figures




read the original abstract
The maximum parsimony phylogenetic tree reconstruction problem is NP-hard, presenting a computational bottleneck for classical computing and motivating the exploration of emerging paradigms like quantum computing. To this end, we design three optimization models compatible with both classical and quantum solvers. Our method directly searches the complete solution space of all possible tree topologies and ancestral states, thereby avoiding the potential biases associated with pre-constructing candidate internal nodes. Among these models, the branch-based model drastically reduces the number of variables and explicit constraints through a specific variable definition, providing a novel modeling approach effective not only for phylogenetic tree building but also for other tree problems. The correctness of this model is validated with a classical solver, which obtains solutions that are generally better than those from heuristics on the GAPDH gene dataset. Moreover, our quantum simulations successfully find the exact optimal solutions for small-scale instances with rapid convergence, highlighting the potential of quantum computing to offer a new avenue for solving these intractable problems in evolutionary biology.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces three optimization models for maximum parsimony phylogenetic tree reconstruction that are compatible with both classical and quantum solvers. The models directly enumerate the space of all tree topologies and ancestral states rather than pre-constructing candidate nodes. A novel branch-based model is presented that uses a specific variable definition to reduce the number of variables and explicit constraints. Classical solver results on the GAPDH gene dataset are reported to outperform standard heuristics, while quantum simulations are shown to recover exact optima for small instances with rapid convergence.
Significance. If the branch-based formulation is shown to be exactly equivalent to the standard maximum-parsimony objective, the modeling approach could provide a reusable template for other tree-structured combinatorial problems. The direct-search strategy avoids certain heuristic biases, and the reported classical outperformance on a real gene dataset plus the small-scale quantum results together indicate a plausible route for quantum methods in phylogenetics once larger instances become tractable.
major comments (2)
- The central claim that the branch-based variable definition exactly encodes every valid tree topology and all parsimony change counts without hidden restrictions or omissions is load-bearing for both the GAPDH results and the quantum simulations. No formal equivalence proof or exhaustive enumeration check is supplied to confirm that every Steiner tree and every ancestral-state assignment remains reachable under the reduced variable set.
- Validation section: the statement that the classical solver obtains solutions 'generally better' than heuristics on the GAPDH dataset lacks a full dataset description, an explicit error analysis, and a statistical comparison that controls for implementation details of the heuristic baselines. Without these, it is impossible to determine whether the reported advantage is attributable to the model or to solver-specific tuning.
minor comments (2)
- Notation for the three models is introduced without a consolidated table comparing variable counts, constraint counts, and objective functions; such a table would improve readability.
- The quantum simulation results are presented only for 'small-scale instances'; the precise instance sizes (number of taxa and characters) should be stated explicitly in the main text rather than relegated to supplementary material.
Simulated Author's Rebuttal
We are grateful to the referee for the thorough review and constructive feedback on our manuscript. We respond to the major comments point by point and indicate the revisions we will make to address them.
read point-by-point responses
-
Referee: The central claim that the branch-based variable definition exactly encodes every valid tree topology and all parsimony change counts without hidden restrictions or omissions is load-bearing for both the GAPDH results and the quantum simulations. No formal equivalence proof or exhaustive enumeration check is supplied to confirm that every Steiner tree and every ancestral-state assignment remains reachable under the reduced variable set.
Authors: We agree that a formal proof of equivalence would strengthen the manuscript. In the revised version, we will add a dedicated subsection providing a rigorous proof that the branch-based formulation is exactly equivalent to the standard maximum-parsimony objective. The proof will show that every valid tree topology and ancestral-state assignment is representable under the reduced variable set, with no hidden restrictions or omissions, and that the objective correctly counts all changes. We will also include exhaustive enumeration verification on small instances to confirm completeness. revision: yes
-
Referee: Validation section: the statement that the classical solver obtains solutions 'generally better' than heuristics on the GAPDH dataset lacks a full dataset description, an explicit error analysis, and a statistical comparison that controls for implementation details of the heuristic baselines. Without these, it is impossible to determine whether the reported advantage is attributable to the model or to solver-specific tuning.
Authors: We acknowledge that the current validation would benefit from greater detail and rigor. In the revision, we will expand the GAPDH dataset description to include its source, number of taxa, alignment length, and preprocessing steps. We will add an explicit error analysis and include statistical comparisons (such as paired Wilcoxon signed-rank tests) against the heuristic baselines, with full reporting of implementation parameters to control for tuning effects and ensure the advantage is attributable to the model. revision: yes
Circularity Check
No significant circularity; models encode parsimony from first principles and are externally validated
full rationale
The paper formulates three optimization models that directly encode the maximum parsimony objective over the full space of tree topologies and ancestral states, without reducing any claimed result to a fitted parameter or self-citation. The branch-based model's variable definition is presented as a modeling choice that reduces variables while preserving equivalence to the standard criterion; correctness is checked by running a classical solver on the external GAPDH gene dataset and comparing against heuristics, plus quantum simulations on small instances. No load-bearing self-citations, uniqueness theorems from prior author work, or ansatz smuggling appear in the provided derivation chain. The central claims rest on explicit model construction plus independent solver output rather than tautological re-derivation of inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Maximum parsimony phylogenetic tree reconstruction is NP-hard
Reference graph
Works this paper leans on
-
[1]
Depth-based model The primary challenge in modeling an unrooted tree is to impose a coherent structure that prevents cycles. A common and intuitive strategy is to establish a hier- archy by defining the depth of each node relative to the reference node, and to use constraints to ensure all con- nections flow in a single direction, thereby obtaining an acy...
-
[2]
Position-based model As an alternative to the depth-based method, we can assign positions to the nodes. Since leaf nodes have only 4 a single incoming edge, their positions do not need to be assigned as variables, which significantly reduces the total number of variables required. In the position-based model, we assign each non- reference internal node to...
-
[3]
Observing these challenges, we further propose a highly simplified branch- based model
Branch-based model The depth-based model requires too many variables and constraints, while the objective function of the position-based model is overly complex. Observing these challenges, we further propose a highly simplified branch- based model. Since the internal nodes are essentially identical before the base or sequence information is determined, w...
-
[4]
Felsenstein, in Inferring phylogenies (2004), pp
J. Felsenstein, in Inferring phylogenies (2004), pp. 664– 664
work page 2004
-
[5]
J. Bull and H. Wichman, Annual Review of Ecology and systematics 32, 183 (2001)
work page 2001
-
[6]
T. J. Davies, S. A. Fritz, R. Grenyer, C. D. L. Orme, J. Bielby, O. R. Bininda-Emonds, M. Cardillo, K. E. Jones, J. L. Gittleman, G. M. Mace, et al., Proceedings of the National Academy of Sciences 105, 11556 (2008)
work page 2008
-
[7]
N. O’Donoghue and R. Yordanova, Trakia Journal of Sci- ences 18, 118 (2020)
work page 2020
-
[8]
C. H. Saslis-Lagoudakis, B. B. Klitgaard, F. Forest, L. Francis, V. Savolainen, E. M. Williamson, and J. A. Hawkins, PloS one 6, e22275 (2011)
work page 2011
-
[9]
W. Cancino and A. C. B. Delbem, New Achievements in Evolutionary Computation pp. 135–156 (2010)
work page 2010
-
[10]
M. N. Puttick, J. E. O’Reilly, D. Pisani, and P. C. Donoghue, Palaeontology 62, 1 (2019)
work page 2019
-
[11]
W. H. Day, D. S. Johnson, and D. Sankoff, Mathematical biosciences 81, 33 (1986)
work page 1986
- [12]
-
[13]
B. Kirkup and J. Kim, Unpublished manuscript. Depart- ment of Ecology and Evolutionary Biology, Yale Univer- sity, USA 26 (2000)
work page 2000
-
[14]
P. W. Shor, SIAM review 41, 303 (1999)
work page 1999
-
[15]
Li, arXiv preprint arXiv:2307.12492 (2023)
J. Li, arXiv preprint arXiv:2307.12492 (2023)
- [16]
-
[17]
F. Gemeinhardt, A. Garmendia, M. Wimmer, B. Weder, and F. Leymann, ACM Computing Surveys 56, 1 (2023)
work page 2023
-
[18]
L. R. Foulds and R. L. Graham, Advances in Applied mathematics 3, 43 (1982)
work page 1982
-
[19]
F. K. Hwang, D. S. Richards, and P. Winter, North- Holland, Amsterdam 1, 3 (1992)
work page 1992
-
[20]
D. Catanzaro, R. Ravi, and R. Schwartz, Algorithms for Molecular Biology 8, 1 (2013)
work page 2013
-
[21]
H. H. Bach, D. K. Nguyen, and N. N. V. Dung, in Inter- national Conference on Future Data and Security Engi- neering (Springer, 2024), pp. 158–170
work page 2024
-
[22]
M. D. Hendy and D. Penny, Mathematical biosciences 59, 277 (1982)
work page 1982
-
[23]
L. Kannan and W. C. Wheeler, Algorithms for molecular biology 7, 1 (2012)
work page 2012
-
[24]
W. M. Brown, E. M. Prager, A. Wang, and A. C. Wilson, Journal of molecular evolution 18, 225 (1982)
work page 1982
-
[25]
Wakeley, Molecular Biology and Evolution 11, 436 (1994)
J. Wakeley, Molecular Biology and Evolution 11, 436 (1994)
work page 1994
-
[26]
L. Perron and F. Didier, Cp-sat, URL https: //developers.google.com/optimization/cp/cp_ solver/
- [27]
-
[28]
J. R. McClean, J. Romero, R. Babbush, and A. Aspuru- Guzik, New Journal of Physics 18, 023023 (2016)
work page 2016
-
[29]
A Quantum Approximate Optimization Algorithm
E. Farhi, J. Goldstone, and S. Gutmann, arXiv preprint arXiv:1411.4028 (2014)
work page internal anchor Pith review Pith/arXiv arXiv 2014
-
[30]
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’brien, Nature communications 5, 4213 (2014)
work page 2014
-
[31]
A. Javadi-Abhari, M. Treinish, K. Krsulich, C. J. Wood, J. Lishman, J. Gacon, S. Martiel, P. D. Nation, L. S. Bishop, A. W. Cross, et al., Quantum computing with Qiskit (2024), 2405.08810
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[32]
PennyLane: Automatic differentiation of hybrid quantum-classical computations
V. Bergholm, J. Izaac, M. Schuld, C. Gogolin, S. Ahmed, V. Ajith, M. S. Alam, G. Alonso-Linaje, B. AkashNarayanan, A. Asadi, et al., arXiv preprint arXiv:1811.04968 (2018)
work page internal anchor Pith review Pith/arXiv arXiv 2018
- [33]
-
[34]
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, nature 549, 242 (2017). 10
work page 2017
- [35]
- [36]
- [37]
- [38]
- [39]
-
[40]
W. M. Fitch, Systematic Biology 20, 406 (1971)
work page 1971
-
[41]
Felsenstein, Systematic zoology 27, 401 (1978)
J. Felsenstein, Systematic zoology 27, 401 (1978)
work page 1978
-
[42]
Sankoff, SIAM Journal on Applied Mathematics 28, 35 (1975)
D. Sankoff, SIAM Journal on Applied Mathematics 28, 35 (1975)
work page 1975
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.