AiiDA-TrainsPot: Towards automated training of neural-network interatomic potentials
Pith reviewed 2026-05-18 17:04 UTC · model grok-4.3
The pith
An automated workflow calibrates committee disagreement on the fly to select ab initio calculations reliably when training neural-network interatomic potentials.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
AiiDA-TrainsPot automates the creation of neural-network interatomic potentials by linking DFT computations with active learning. The key advance is on-the-fly calibration of committee disagreement against ab initio reference errors, which yields reliable uncertainty estimates. This calibrated measure, validated through electronic-structure descriptors and dimensionality reduction, minimizes both false positives and false negatives when choosing structures for further first-principles evaluation across the tested carbon and alloy systems.
What carries the argument
On-the-fly calibration of committee disagreement against ab initio reference errors, which supplies reliable uncertainty estimates that guide active-learning decisions on which structures to compute from first principles.
If this is right
- The same calibrated criterion supports both training potentials from scratch and fine-tuning foundation models.
- The workflow successfully handles pristine, defective, and amorphous carbon structures as well as alloy phase transitions.
- Modular design allows swapping in different neural-network interatomic potential backends without rewriting the active-learning loop.
- On-the-fly calibration reduces the number of expensive ab initio calculations needed while maintaining coverage of relevant configurations.
Where Pith is reading between the lines
- The calibration approach could be tested on systems outside the demonstrated carbon and dichalcogenide cases to check transferability of the uncertainty estimates.
- Lower false-negative rates might allow safer use of the resulting potentials in long molecular-dynamics runs where rare events matter.
- Because the workflow is built on an existing automation platform, it could be combined with high-throughput material-screening campaigns that already generate large structure databases.
Load-bearing premise
Electronic-structure descriptors combined with dimensionality reduction can reliably validate the calibrated committee disagreement criterion without introducing selection bias in the active-learning loop.
What would settle it
Apply the workflow to a new material system, collect the structures it selects versus those it skips, and check whether many skipped structures later show large actual errors when computed ab initio or whether many selected structures prove unnecessary.
Figures






read the original abstract
Crafting neural-network interatomic potentials (NNIPs) remains a complex task, demanding specialized expertise in both machine learning and electronic-structure calculations. Here, we introduce AiiDA-TrainsPot, an automated, open-source, and user-friendly workflow that streamlines the creation of accurate NNIPs by orchestrating density-functional-theory calculations, data augmentation strategies, and classical molecular dynamics. Our active-learning strategy leverages on-the-fly calibration of committee disagreement against ab initio reference errors to ensure reliable uncertainty estimates. We use electronic-structure descriptors and dimensionality reduction to analyze the efficiency of this calibrated criterion, and show that it minimizes both false positives and false negatives when deciding what to compute from first principles. AiiDA-TrainsPot has a modular design that supports multiple NNIP backends, enabling both the training of NNIPs from scratch and the fine-tuning of foundation models. We demonstrate its capabilities through automated training campaigns targeting pristine and defective carbon allotropes, including amorphous carbon, as well as structural phase transitions in monolayer $\mathrm{W_xMo_{1-x}Te_2}$ alloys.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces AiiDA-TrainsPot, an automated open-source workflow built on AiiDA for training neural-network interatomic potentials (NNIPs). It integrates DFT calculations, data augmentation, and molecular dynamics, with a core active-learning loop that performs on-the-fly calibration of committee disagreement against ab initio reference errors to produce uncertainty estimates. Electronic-structure descriptors combined with dimensionality reduction are used to analyze the efficiency of this calibrated criterion on carbon allotropes (including amorphous carbon) and WxMo1-xTe2 alloy systems, with the claim that the approach minimizes both false positives and false negatives when selecting structures for first-principles evaluation. The workflow is modular and supports training from scratch as well as fine-tuning of foundation models.
Significance. If the on-the-fly calibration produces genuinely independent and reliable uncertainty estimates that generalize beyond the training data, the work would meaningfully advance automated, reproducible NNIP development for complex materials systems. The demonstrations on defective carbon and alloy phase transitions, together with support for multiple backends, would provide practical value for reducing expert intervention in potential training campaigns.
major comments (1)
- [Results section on descriptor-based analysis of the calibrated criterion] The section describing the analysis of the calibrated committee disagreement (via electronic-structure descriptors and dimensionality reduction) must explicitly state whether this validation is performed on a fully independent hold-out set of structures or on configurations already visited or selected by the active-learning procedure itself. If the reduced descriptor space is constructed from the same committee evaluations used for calibration, the reported minimization of false positives and false negatives risks selection bias and does not independently confirm the reliability of the uncertainty estimates.
minor comments (2)
- [Abstract] The abstract states that the calibrated criterion 'minimizes both false positives and false negatives' but provides no quantitative thresholds, error metrics, or cross-validation details; a brief clarification of the decision rule and evaluation protocol would improve clarity.
- [Methods] Ensure that the precise definition of the committee (number of models, training protocol, and disagreement metric) is given in a dedicated methods subsection or table so that the on-the-fly calibration procedure can be reproduced without ambiguity.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive comment on the descriptor-based analysis. We have revised the relevant section to explicitly address the nature of the dataset used and the implications for interpreting the results.
read point-by-point responses
-
Referee: [Results section on descriptor-based analysis of the calibrated criterion] The section describing the analysis of the calibrated committee disagreement (via electronic-structure descriptors and dimensionality reduction) must explicitly state whether this validation is performed on a fully independent hold-out set of structures or on configurations already visited or selected by the active-learning procedure itself. If the reduced descriptor space is constructed from the same committee evaluations used for calibration, the reported minimization of false positives and false negatives risks selection bias and does not independently confirm the reliability of the uncertainty estimates.
Authors: We thank the referee for this observation. The analysis presented in the Results section was performed on configurations visited or selected during the active-learning procedure itself, as the on-the-fly calibration of committee disagreement is an integral part of the workflow and occurs on the structures encountered in each training campaign. The reduced descriptor space is therefore constructed from the same set of committee evaluations used for calibration. We agree that this constitutes an in-sample analysis and carries a risk of selection bias; it does not provide an independent confirmation of the uncertainty estimates on fully unseen data. The intent of the section is to demonstrate the practical efficiency of the calibrated criterion in minimizing false positives and false negatives within the automated campaigns on carbon allotropes and the alloy systems, rather than to claim external validation. In the revised manuscript we have added explicit statements clarifying the in-sample nature of the analysis and discussing its limitations for assessing generalization. We believe this improves transparency without altering the reported findings. revision: yes
Circularity Check
No significant circularity in the presented workflow or validation chain.
full rationale
The abstract describes an on-the-fly calibration of committee disagreement against ab initio reference errors as part of the active-learning strategy, followed by separate analysis using electronic-structure descriptors and dimensionality reduction to assess efficiency on carbon allotropes and alloy systems. No equations or explicit statements in the provided text demonstrate that calibration parameters or decision thresholds are fitted directly to the same data used for the final NNIP training or that the descriptor-based validation reduces to the active-learning selections by construction. The workflow is presented as modular with demonstrations on specific targets, and the analysis functions as an external check rather than a self-referential loop. No load-bearing self-citations, imported uniqueness theorems, or ansatz smuggling are evident. The derivation chain remains self-contained against the described benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption DFT calculations provide reliable reference errors for calibrating committee disagreement.
- domain assumption Committee disagreement can be mapped to actual prediction errors via on-the-fly calibration.
Forward citations
Cited by 1 Pith paper
-
Spin Dynamics from Atomistic Quantum Simulations
Kubo theory yields T1 and T2 from spin-lattice correlation functions; these are evaluated with ML-driven ab initio molecular dynamics and match experimental T1 values for the NV center in diamond.
Reference graph
Works this paper leans on
-
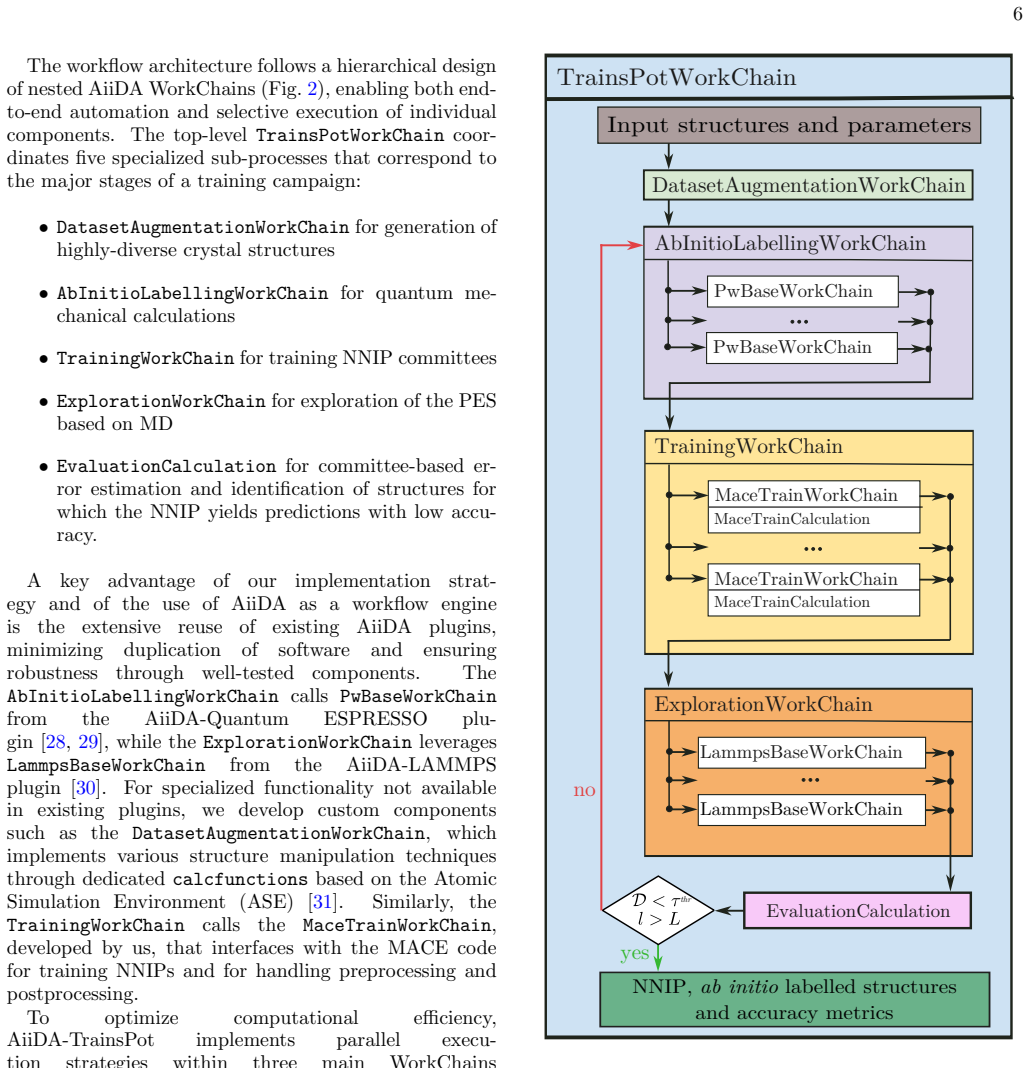
[1]
open), cell parameters, atomic species and atomic positions
Input structures AiiDA-TrainsPot can start from a small set of ini- tial atomistic structures{X } (0), determined by boundary conditions (periodic vs. open), cell parameters, atomic species and atomic positions. The number and diversity of input structures should reflect the target applications: for example, the study of temperature-dependent prop- erties...
-
[2]
Dataset augmentation In the dataset augmentation stage, additional struc- tures are generated by manipulating the initial set {X }(0). All manipulations can be controlled through cus- tomizable parameters to tailor the augmentation process according to specific user needs; we group them in the following categories: •Supercells: Initial structures are repl...
-
[3]
Ab Initio Labelling After the data augmentation stage, AiiDA-TrainsPot starts the active learning loop, which is represented by the orange circle in Fig. 1. Each structureX i in the augmented dataset is labeled through DFT calcula- tions to obtain high-fidelity reference values for energies, forces, and stress tensors. We use the compact notation LDF T (X...
-
[4]
Training neural-network interatomic potentials The labeled dataset Xi,L DF T (Xi) (1) is used to train a committee ofMNNIPs{Φ j}M j=1, each with identical architecture but initialized with different random seeds. Prior to training, all structures are systematically par- titioned into three subsets, ensuring representative sam- pling across different struc...
-
[5]
Exploration by molecular dynamics After a committee of NNIPs is trained, the workflow employs MD simulations to systematically explore the potential energy landscape. This exploration phase is critical for identifying configurations where the NNIPs might have insufficient accuracy, thus guiding the selec- tion of additional structures forab initiocalculat...
-
[6]
Committee Evaluation This stage aims at identifying structures that are poorly predicted by the NNIPs; those are good candi- dates to be labeled withab initiocalculations and in- cluded in the training dataset. However, while Bayesian neural networks (NNs) come with a well-defined proba- bilistic uncertainty quantification, no such Bayesian error estimati...
-
[7]
to 9,537 structures in the final iteration. For energy and stress tensor components, we observe a consistent decrease in prediction errors as the active learning pro- gresses. Interestingly, errors on forces increase from the first to the second iteration, and then decrease monoton- ically for all the following iterations, suggesting that the active learn...
-
[8]
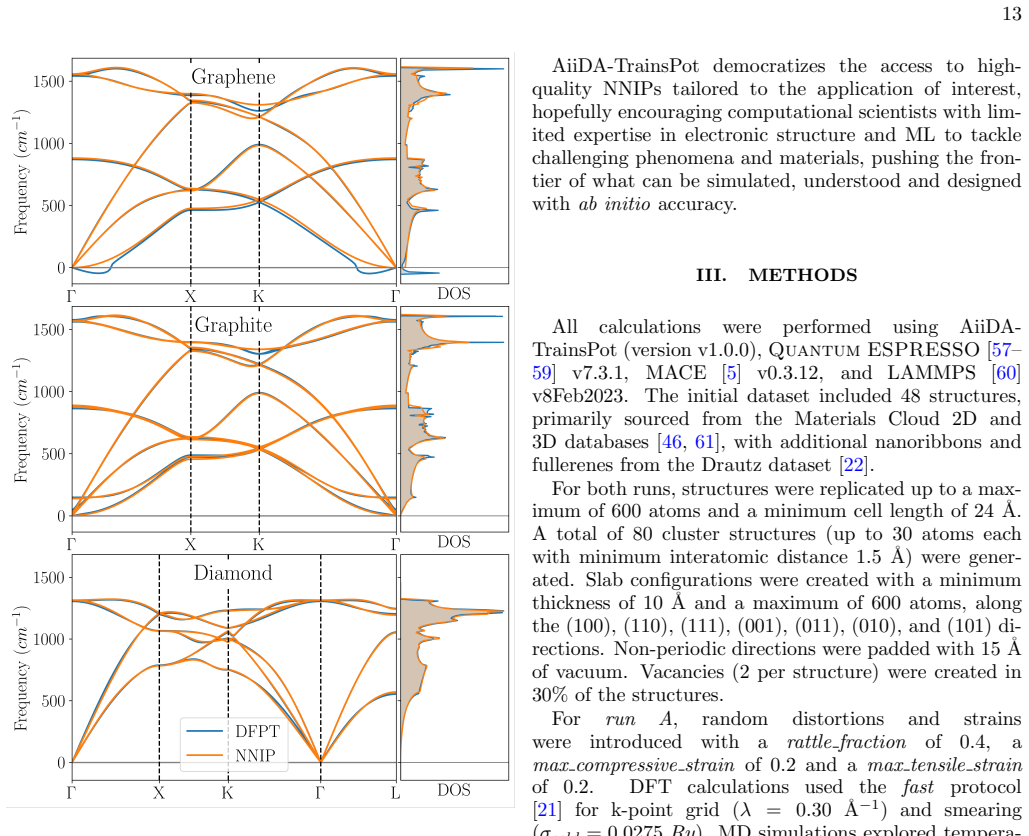
or DFT (row 2). The third and fourth rows correspond to structures relaxed with DFT, with subsequent energy evalu- ation using the NNIP (row 3) or DFT (row 4). To assess the transferability of our NNIP to defective structures, we compute the formation energies of three representative point defects in graphene: the monova- cancy, divacancy, and Stone-Wales...
-
[9]
MD simulations explored tempera- tures ranging from 0 to 5000 K and pressures from−5 to 5 kbar
for k-point grid (λ= 0.30 ˚A−1) and smearing (σcold = 0.0275Ry). MD simulations explored tempera- tures ranging from 0 to 5000 K and pressures from−5 to 5 kbar. Forrun B, dataset augmentation parameters were op- timized for near-equilibrium conditions:rattle fraction was reduced to 0.3, while strain ranges were increased tomax compressive strainof 0.3 and...
-
[10]
MaX - Materials De- sign at the Exascale
in LAMMPS. Active learning thresholds on energy, forces, and stress tensor were set to 2 meV, 50 meV/ ˚A, 14 10 meV/˚A3, respectively, with a maximum of 1000 struc- tures selected per iteration. IV. DATA AVAILABILITY The training datasets and trained models are available on the Materials Cloud Archive [63]. V. CODE AVAILABILITY AiiDA-TrainsPot is availabl...
- [11]
-
[12]
V. L. Deringer, M. A. Caro, and G. Cs´ anyi, Ma- chine Learning Interatomic Potentials as Emerging Tools for Materials Science, Advanced Materials31, 1902765 (2019)
work page 2019
-
[13]
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nature Communications13, 2453 (2022)
work page 2022
-
[14]
A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth, and B. Kozinsky, Learning local equivariant representations for large-scale atomistic dy- namics, Nature Communications14, 579 (2023)
work page 2023
-
[15]
I. Batatia, D. P. Kovacs, G. N. C. Simm, C. Ortner, and G. Csanyi, MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Molec- ular Force Fields, Advances in Neural Information Pro- cessing Systems35, 11423 (2022)
work page 2022
- [16]
-
[17]
A. Mazitov, F. Bigi, M. Kellner, P. Pegolo, D. Tisi, G. Fraux, S. Pozdnyakov, P. Loche, and M. Ceri- otti, PET-MAD, a lightweight universal interatomic potential for advanced materials modeling (2025), arXiv:2503.14118 [cond-mat.mtrl-sci]
- [18]
-
[19]
K. T. Sch¨ utt, P. Kessel, M. Gastegger, K. A. Nicoli, A. Tkatchenko, and K.-R. M¨ uller, SchNetPack: A Deep Learning Toolbox For Atomistic Systems, Journal of Chemical Theory and Computation15, 448–455 (2018)
work page 2018
-
[20]
L. Talirz, S. Kumbhar, E. Passaro, A. V. Yakutovich, V. Granata, F. Gargiulo, M. Borelli, M. Uhrin, S. P. Huber, S. Zoupanos, C. S. Adorf, C. W. Andersen, O. Sch¨ utt, C. A. Pignedoli, D. Passerone, J. VandeVon- dele, T. C. Schulthess, B. Smit, G. Pizzi, and N. Marzari, Materials Cloud, a platform for open computational sci- ence, Scientific Data7, 299 (2020)
work page 2020
-
[21]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, and K. A. Persson, The Materials Project: A materials genome approach to accelerating materials innovation, APL Materials1, 011002 (2013)
work page 2013
-
[22]
G. Bergerhoff, R. Hundt, R. Sievers, and I. D. Brown, The inorganic crystal structure data base, Journal of Chemical Information and Computer Sciences23, 66 (1983)
work page 1983
-
[23]
S. Graˇ zulis, D. Chateigner, R. T. Downs, A. F. T. Yokochi, M. Quir´ os, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck, and A. Le Bail, Crystallogra- phy Open Database—an open-access collection of crystal structures, Journal of Applied Crystallography42, 726 (2009). 15
work page 2009
-
[24]
S. Graˇ zulis, A. Daˇ skeviˇ c, A. Merkys, D. Chateigner, L. Lutterotti, M. Quir´ os, N. R. Serebryanaya, P. Moeck, R. T. Downs, and A. Le Bail, Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration, Nucleic Acids Research40, D420 (2011)
work page 2011
-
[25]
P. Villars, N. Onodera, and S. Iwata, The linus pauling file (lpf) and its application to materials design, Journal of Alloys and Compounds279, 1 (1998)
work page 1998
-
[26]
P. Villars, M. Berndt, K. Brandenburg, K. Cenzual, J. Daams, F. Hulliger, T. Massalski, H. Okamoto, K. Os- aki, A. Prince, H. Putz, and S. Iwata, The pauling file, binaries edition, Journal of Alloys and Compounds367, 293 (2004)
work page 2004
-
[27]
E. Blokhin and P. Villars, The PAULING FILE Project and Materials Platform for Data Science: From Big Data Toward Materials Genome, inHandbook of Materials Modeling : Methods: Theory and Modeling, edited by W. Andreoni and S. Yip (Springer International Pub- lishing, Cham, 2018) pp. 1–26
work page 2018
-
[28]
G. Prandini, A. Marrazzo, I. E. Castelli, N. Mounet, and N. Marzari, Precision and efficiency in solid-state pseu- dopotential calculations, npj Computational Materials4, 72 (2018)
work page 2018
-
[29]
P. E. Bl¨ ochl, Projector augmented-wave method, Phys. Rev. B50, 17953 (1994)
work page 1994
-
[30]
A. Dal Corso, Pseudopotentials periodic table: From H to Pu, Computational Materials Science95, 337 (2014)
work page 2014
- [31]
- [32]
-
[33]
S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, Journal of Computational Chemistry27, 1787 (2006)
work page 2006
-
[34]
M. Abdar, F. Pourpanah, S. Hussain, D. Rezazadegan, L. Liu, M. Ghavamzadeh, P. Fieguth, X. Cao, A. Khos- ravi, U. R. Acharya, V. Makarenkov, and S. Nahavandi, A review of uncertainty quantification in deep learning: Techniques, applications and challenges, Information Fu- sion76, 243 (2021)
work page 2021
-
[35]
J. Behler, Representing potential energy surfaces by high-dimensional neural network potentials, Journal of Physics: Condensed Matter26, 183001 (2014)
work page 2014
-
[36]
L. Chen, I. Sukuba, M. Probst, and A. Kaiser, Itera- tive training set refinement enables reactive molecular dynamicsviamachine learned forces, RSC Advances10, 4293 (2020)
work page 2020
-
[37]
L. Kahle and F. Zipoli, Quality of uncertainty estimates from neural network potential ensembles, Physical Re- view E105, 015311 (2022)
work page 2022
-
[38]
S. P. Huber, S. Zoupanos, M. Uhrin, L. Talirz, L. Kahle, R. H¨ auselmann, D. Gresch, T. M¨ uller, A. V. Yakutovich, C. W. Andersen, F. F. Ramirez, C. S. Adorf, F. Gargiulo, S. Kumbhar, E. Passaro, C. Johnston, A. Merkys, A. Ce- pellotti, N. Mounet, N. Marzari, B. Kozinsky, and G. Pizzi, AiiDA 1.0, a scalable computational infras- tructure for automated re...
work page 2020
- [39]
-
[40]
Theaiida-lammpsplugin is available athttps:// github.com/aiidaplugins/aiida-lammps
-
[41]
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Pe- terson, C. Rostgaard, J. Schiøtz, O. Sch...
work page 2017
- [42]
- [43]
-
[44]
B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson, and T. L. Windus, New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Commu- nity, Journal of Chemical Information and Modeling59, 4814 (2019)
work page 2019
-
[45]
D. Feller, The role of databases in support of computa- tional chemistry calculations, Journal of Computational Chemistry17, 1571 (1996)
work page 1996
-
[46]
K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li, and T. L. Windus, Ba- sis Set Exchange: A Community Database for Compu- tational Sciences, Journal of Chemical Information and Modeling47, 1045 (2007)
work page 2007
-
[47]
B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, and P. Widmark, Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set, The Journal of Physical Chemistry A108, 2851 (2004)
work page 2004
-
[48]
B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, and P. Widmark, New Relativistic ANO Basis Sets for Tran- sition Metal Atoms, The Journal of Physical Chemistry A109, 6575 (2005)
work page 2005
-
[49]
B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, and P. Widmark, New relativistic ANO basis sets for actinide atoms, Chemical Physics Letters409, 295 (2005)
work page 2005
-
[50]
B. O. Roos, R. Lindh, P. Malmqvist, V. Veryazov, P. Widmark, and A. C. Borin, New Relativistic Atomic Natural Orbital Basis Sets for Lanthanide Atoms with Applications to the Ce Diatom and LuF3, The Journal of Physical Chemistry A112, 11431 (2008)
work page 2008
-
[51]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett. 77, 3865 (1996)
work page 1996
-
[52]
T. W. Ko, J. A. Finkler, S. Goedecker, and J. Behler, A fourth-generation high-dimensional neural network po- tential with accurate electrostatics including non-local charge transfer, Nature Communications12, 398 (2021)
work page 2021
-
[53]
F. Banhart, J. Kotakoski, and A. V. Krasheninnikov, Structural Defects in Graphene, ACS Nano5, 26 (2011)
work page 2011
-
[54]
J. J. Palacios, J. Fern´ andez-Rossier, and L. Brey, Vacancy-induced magnetism in graphene and graphene ribbons, Physical Review B77, 195428 (2008). 16
work page 2008
- [55]
- [56]
-
[57]
A. Laio and M. Parrinello, Escaping free-energy min- ima, Proceedings of the National Academy of Sciences 99, 12562 (2002)
work page 2002
-
[58]
M. Bonomi, D. Branduardi, G. Bussi, C. Camil- loni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia, and M. Parrinello, PLUMED: A portable plugin for free-energy calculations with molec- ular dynamics, Computer Physics Communications180, 1961 (2009)
work page 1961
-
[59]
G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni, and G. Bussi, PLUMED 2: New feathers for an old bird, Computer Physics Communications185, 604 (2014)
work page 2014
-
[60]
The PLUMED consortium, Promoting transparency and reproducibility in enhanced molecular simulations, Na- ture Methods16, 670–673 (2019)
work page 2019
-
[61]
S. P. Huber, E. Bosoni, M. Bercx, J. Br¨ oder, A. De- gomme, V. Dikan, K. Eimre, E. Flage-Larsen, A. Gar- cia, L. Genovese, D. Gresch, C. Johnston, G. Petretto, S. Ponc´ e, G.-M. Rignanese, C. J. Sewell, B. Smit, V. Tse- plyaev, M. Uhrin, D. Wortmann, A. V. Yakutovich, A. Zadoks, P. Zarabadi-Poor, B. Zhu, N. Marzari, and G. Pizzi, Common workflows for com...
work page 2021
- [62]
- [63]
- [64]
-
[65]
L. Bastonero, C. Malica, E. Macke, M. Bercx, S. Hu- ber, I. Timrov, and N. Marzari, First-principles Hubbard parameters with automated and reproducible workflows, npj Computational Materials11, 183 (2025)
work page 2025
- [66]
-
[67]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococ- cioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. S...
work page 2009
-
[68]
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavaz- zoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carn- imeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Ko...
work page 2017
-
[69]
P. Giannozzi, O. Baseggio, P. Bonf` a, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru, and S. Baroni, Quantum ESPRESSO toward the exascale, The Journal of Chemical Physics152, 154105 (2020)
work page 2020
-
[70]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS—a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Commun...
work page 2022
- [71]
-
[72]
Y. Zhou, W. A. Saidi, and K. A. Fichthorn, A Force Field for Describing the Polyvinylpyrrolidone-Mediated Solution-Phase Synthesis of Shape-Selective Ag Nanopar- ticles, The Journal of Physical Chemistry C118, 3366 (2014)
work page 2014
-
[73]
D. Bidoggia, N. Manko, M. Peressi, and A. Marrazzo, Automated training of neural-network interatomic poten- tials, 10.24435/materialscloud:8d-kj (2025)
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.