Benchmarking thermostat algorithms in molecular dynamics simulations of a binary Lennard-Jones glass-former model
Pith reviewed 2026-05-18 17:00 UTC · model grok-4.3
The pith
The Grønbech-Jensen--Farago Langevin thermostat provides the most consistent sampling of temperature and potential energy in molecular dynamics of a binary Lennard-Jones glass former.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
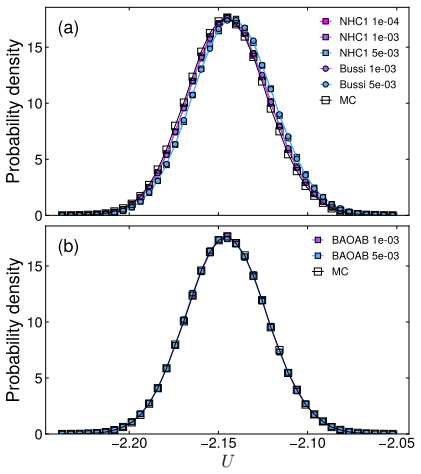
Among the Langevin methods, the Grønbech-Jensen--Farago scheme provided the most consistent sampling of both temperature and potential energy. The Nosé-Hoover chain and Bussi thermostats offer reliable temperature control but show time-step dependence in potential energy. Langevin dynamics incurs roughly twice the computational cost from random number generation and causes diffusion coefficients to decrease with higher friction. This benchmarking on the binary Lennard-Jones model supplies practical advice for thermostat selection in classical molecular dynamics.
What carries the argument
Systematic benchmarking of thermostat algorithms on the binary Lennard-Jones glass-former model by monitoring time-step dependence in sampled temperature, potential energy, and diffusion.
If this is right
- If the claim holds, simulations using the Grønbech-Jensen--Farago thermostat will yield potential energy values independent of the chosen time step.
- Langevin-based simulations will require accounting for increased computational overhead due to random number generation.
- Diffusion-related properties calculated from Langevin dynamics will need correction for the friction-induced reduction.
- Temperature control with Nosé-Hoover or Bussi methods will remain reliable but potential energy accuracy will depend on using sufficiently small time steps.
Where Pith is reading between the lines
- The relative performance of these thermostats may generalize to other model systems used in materials simulations.
- Further tests could examine how these thermostats affect computed quantities like viscosity or specific heat in the glass former.
- Applying the same comparison to ab initio molecular dynamics or machine-learning potentials could extend the guidance to more complex systems.
Load-bearing premise
The binary Lennard-Jones glass-former model and the selected observables such as velocities and potential energy are representative enough to expose general differences in how thermostats perform across various molecular systems.
What would settle it
A new set of simulations on the same model but with a different thermostat showing no time-step dependence in potential energy comparable to or better than the Grønbech-Jensen--Farago scheme would falsify its superiority in consistency.
Figures












read the original abstract
A systematic comparison was carried out to assess the influence of representative thermostat methods in constant-temperature molecular dynamics simulations. The thermostat schemes considered include the Nos\'e--Hoover thermostat and its chain generalisation, the Bussi velocity rescaling method, and several implementations of the Langevin dynamics. Using a binary Lennard-Jones liquid as a model glass former, we investigated how the sampling of physical observables, such as particle velocities and potential energy, responds to changes in time step across these thermostats. While the Nos\'e--Hoover chain and Bussi thermostats provide reliable temperature control, a pronounced time-step dependence was observed in the potential energy. Amongst the Langevin methods, the Gr{\o}nbech-Jensen--Farago scheme provided the most consistent sampling of both temperature and potential energy. Nonetheless, Langevin dynamics typically incurs approximately twice the computational cost due to the overhead of random number generation, and exhibits a systematic decrease in diffusion coefficients with increasing friction. This study presents a broad comparison of thermostat methods using a binary Lennard-Jones glass-former model, offering practical guidance for the choice of thermostats in classical molecular dynamics simulations. These findings provide useful insights for diverse applications, including glass transition, phase separation, and nucleation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. This manuscript reports a benchmark study comparing the performance of various thermostat algorithms—Nosé-Hoover, Nosé-Hoover chains, Bussi velocity rescaling, and multiple Langevin dynamics implementations—in molecular dynamics simulations of a binary Lennard-Jones glass-forming liquid. The authors assess the time-step dependence of key observables including temperature, potential energy, and diffusion coefficients. They conclude that while Nosé-Hoover chain and Bussi methods offer reliable temperature control, potential energy shows pronounced time-step dependence; among Langevin schemes, the Grønbech-Jensen--Farago method exhibits the most consistent sampling, albeit with higher computational cost and friction-dependent diffusion reduction. The study aims to provide practical guidance for thermostat selection in classical MD simulations.
Significance. Should the central findings be confirmed with appropriate statistical controls, the paper would offer useful empirical insights into thermostat performance for simulations of glass-formers, where accurate sampling of energy and dynamics is critical. The broad comparison across methods and the identification of trade-offs (consistency vs. cost) could inform best practices in the molecular dynamics community, particularly for applications involving phase transitions and nucleation.
major comments (1)
- [Results] Results section (Langevin methods comparison): The claim that the Grønbech-Jensen--Farago scheme provides the most consistent sampling of both temperature and potential energy rests on observed weaker time-step dependence, but no statistical uncertainties, error bars, block averages, or autocorrelation times are reported for these quantities. In a glass-former near the transition with long relaxation times, this omission makes it impossible to confirm that the ranking reflects genuine thermostat superiority rather than insufficient sampling or noise, directly affecting the central empirical conclusion.
minor comments (2)
- [Abstract] Abstract: The computational cost comparison (Langevin incurring approximately twice the cost) is stated without reference to the specific hardware or implementation details; move or expand this to the methods or results for clarity.
- [Methods] The manuscript would benefit from explicit statements of the number of independent trajectories, total simulation lengths, and any convergence checks performed on the observables.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive feedback on our benchmarking study. The concern about statistical uncertainties in the Langevin comparison is valid, and we have revised the manuscript to address it directly while preserving the original empirical observations.
read point-by-point responses
-
Referee: [Results] Results section (Langevin methods comparison): The claim that the Grønbech-Jensen--Farago scheme provides the most consistent sampling of both temperature and potential energy rests on observed weaker time-step dependence, but no statistical uncertainties, error bars, block averages, or autocorrelation times are reported for these quantities. In a glass-former near the transition with long relaxation times, this omission makes it impossible to confirm that the ranking reflects genuine thermostat superiority rather than insufficient sampling or noise, directly affecting the central empirical conclusion.
Authors: We appreciate this observation. Our production runs were extended to several hundred relaxation times to account for the slow dynamics near the glass transition, and the weaker time-step dependence for the Grønbech-Jensen-Farago thermostat was reproducible across independent trajectories. Nevertheless, we agree that explicit error quantification strengthens the central claim. In the revised manuscript we have added error bars obtained via block averaging, reported autocorrelation times for temperature and potential energy, and included a brief discussion confirming that the observed ranking remains statistically significant and is not an artifact of noise or inadequate sampling. revision: yes
Circularity Check
Empirical benchmark with no circular derivations or self-referential predictions
full rationale
The paper conducts a direct numerical comparison of thermostat algorithms (Nosé-Hoover, Bussi, Langevin variants including Grønbech-Jensen--Farago) in MD simulations of a binary Lennard-Jones glass former. Observables such as temperature, potential energy, and diffusion are measured across time steps from simulation outputs. No equations derive a result from first principles that reduces to a fitted parameter or self-citation by construction; no predictions are made that are statistically forced by inputs defined within the study. The central claim rests on comparative simulation data rather than any closed logical loop. This is a standard self-contained empirical benchmark, warranting score 0.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The binary Lennard-Jones potential and the chosen system size and cooling protocol produce representative glass-forming behavior for thermostat benchmarking.
Reference graph
Works this paper leans on
-
[1]
M. P. Allen, D. J. Tildesley, Computer Simulation of Liquids, Oxford Uni- versity Press, 2017.doi:10.1093/oso/9780198803195.001.0001
-
[2]
D. Frenkel, B. Smit, Understanding Molecular Simulation, Academic Press, 2023.doi:10.1016/C2009-0-63921-0
-
[3]
M.E.Tuckerman, StatisticalMechanics: TheoryandMolecularSimulation, Oxford University Press, 2023.doi:10.1093/oso/9780198825562.001. 0001. 21
-
[5]
Generalized neural-network representation of high-dimensional potential-energy surfaces
J. Behler, M. Parrinello, Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces, Physical Review Letters 98 (2007) 146401.doi:10.1103/physrevlett.98.146401
-
[6]
L. V. Woodcock, Isothermal molecular dynamics calculations for liq- uid salts, Chemical Physics Letters 10 (1971) 257–261.doi:10.1016/ 0009-2614(71)80281-6
work page 1971
-
[7]
H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola, J. R. Haak, Molecular dynamics with coupling to an external bath, The Journal of Chemical Physics 81 (1984) 3684–3690.doi:10.1063/1.448118
-
[8]
S. Nosé, A unified formulation of the constant temperature molecular dy- namics methods, The Journal of Chemical Physics 81 (1984) 511–519. doi:10.1063/1.447334
-
[9]
S. Nosé, A molecular dynamics method for simulations in the canon- ical ensemble, Molecular Physics 52 (1984) 255–268.doi:10.1080/ 00268978400101201
work page 1984
-
[10]
W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Physical Review A 31 (1985) 1695.doi:10.1103/PhysRevA.31.1695
-
[11]
G. J. Martyna, M. L. Klein, M. Tuckerman, Nosé–Hoover chains: The canonical ensemble via continuous dynamics, The Journal of Chemical Physics 97 (1992) 2635–2643.doi:10.1063/1.463940
-
[12]
T. Schneider, E. Stoll, Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions, Physical Review B 17 (1978) 1302–1322.doi:10.1103/PhysRevB.17.1302
-
[13]
A. Brünger, C. L. Brooks, M. Karplus, Stochastic boundary conditions for molecular dynamics simulations of ST2 water, Chemical Physics Letters 105 (1984) 495–500.doi:10.1016/0009-2614(84)80098-6
-
[14]
E. Vanden-Eijnden, G. Ciccotti, Second-order integrators for Langevin equations with holonomic constraints, Chemical Physics Letters 429 (2006) 310–316.doi:10.1016/j.cplett.2006.07.086
-
[15]
B. Leimkuhler, C. Matthews, Rational Construction of Stochastic Numer- ical Methods for Molecular Sampling, Applied Mathematics Research eX- press 2013 (2012) 34–56.doi:10.1093/amrx/abs010
-
[16]
N. Grønbech-Jensen, O. Farago, A simple and effective Verlet-type algo- rithmforsimulatingLangevindynamics, MolecularPhysics111(2013)983– 991.doi:10.1080/00268976.2012.760055. 22
-
[17]
B.Leimkuhler, C.Matthews, Robustandefficientconfigurationalmolecular sampling via Langevin dynamics, The Journal of Chemical Physics 138 (2013) 174102.doi:10.1063/1.4802990
-
[18]
L. F. Grønbech Jensen, N. Grønbech-Jensen, Accurate configurational and kinetic statistics in discrete-time Langevin systems, Molecular Physics 117 (2019) 2511–2526.doi:10.1080/00268976.2019.1570369
-
[19]
N. Grønbech-Jensen, Complete set of stochastic Verlet-type thermostats for correct Langevin simulations, Molecular Physics 118 (2020) e1662506. doi:10.1080/00268976.2019.1662506
-
[20]
J. Jung, Y. Sugita, Langevin integration for isothermal–isobaric condition with a large time step, The Journal of Chemical Physics 162 (2025) 104108. doi:10.1063/5.0251642
- [21]
-
[22]
G. Bussi, M. Parrinello, Stochastic thermostats: comparison of local and global schemes, Computer Physics Communications 179 (2008) 26–29.doi: 10.1016/j.cpc.2008.01.006
-
[23]
E. Braun, S. M. Moosavi, B. Smit, Anomalous Effects of Velocity Rescaling Algorithms: The Flying Ice Cube Effect Revisited, Journal of Chemical Theory and Computation 14 (2018) 5262–5272.doi:10.1021/acs.jctc. 8b00446
-
[24]
W. Kob, H. C. Andersen, Testing mode-coupling theory for a supercooled binary Lennard-Jones mixture I: The van Hove correlation function, Phys- ical Review E 51 (1995) 4626–4641.doi:10.1103/PhysRevE.51.4626
-
[25]
U. R. Pedersen, T. B. Schrøder, J. C. Dyre, Phase Diagram of Kob- Andersen-Type Binary Lennard-Jones Mixtures, Physical Review Letters 120 (2018) 165501.doi:10.1103/physrevlett.120.165501
-
[26]
P. G. Debenedetti, F. H. Stillinger, Supercooled liquids and the glass tran- sition, Nature 410 (2001) 259–267.doi:10.1038/35065704
-
[27]
T. Gleim, W. Kob, K. Binder, How Does the Relaxation of a Supercooled Liquid Depend on Its Microscopic Dynamics?, Physical Review Letters 81 (1998) 4404.doi:10.1103/PhysRevLett.81.4404
- [28]
-
[29]
L. Berthier, G. Biroli, J.-P. Bouchaud, W. Kob, K. Miyazaki, D. R. Re- ichman, Spontaneous and induced dynamic fluctuations in glass formers. I. General results and dependence on ensemble and dynamics, The Journal of Chemical Physics 126 (2007) 184503.doi:10.1063/1.2721554
-
[30]
L. Berthier, G. Biroli, J.-P. Bouchaud, W. Kob, K. Miyazaki, D. R. Reich- man, Spontaneous and induced dynamic correlations in glass formers. II. Model calculations and comparison to numerical simulations, The Journal of Chemical Physics 126 (2007) 184504.doi:10.1063/1.2721555
-
[31]
D. Coslovich, M. Ozawa, W. Kob, Dynamic and thermodynamic crossover scenarios in the Kob-Andersen mixture: Insights from multi-CPU and multi-GPU simulations, The European Physical Journal E 41 (2018) 62. doi:10.1140/epje/i2018-11671-2
-
[32]
T. B. Schrøder, J. C. Dyre, Solid-like mean-square displacement in glass- forming liquids, The Journal of Chemical Physics 152 (2020) 141101.doi: 10.1063/5.0004093
-
[33]
S. Toxvaerd, J. C. Dyre, Communication: Shifted forces in molecular dynamics, The Journal of Chemical Physics 134 (2011) 081102.doi: 10.1063/1.3558787
-
[34]
R. D. Skeel, J. A. Izaguirre, An impulse integrator for Langevin dynamics, Molecular Physics 100 (2002) 3885–3891.doi:10.1080/ 0026897021000018321
work page 2002
-
[35]
S. Melchionna, Design of quasisymplectic propagators for Langevin dynam- ics, The Journal of Chemical Physics 127 (2007) 044108.doi:10.1063/1. 2753496
work page doi:10.1063/1 2007
-
[36]
doi:10.1007/s10955-023-03104-8
N.Grønbech-Jensen, OntheApplicationofNon-GaussianNoiseinStochas- tic Langevin Simulations, Journal of Statistical Physics 190 (2023) 96. doi:10.1007/s10955-023-03104-8
-
[37]
G. J. Martyna, M. E. Tuckerman, D. J. Tobias, M. L. Klein, Explicit re- versible integrators for extended systems dynamics, Molecular Physics 87 (1996) 1117–1157.doi:10.1080/00268979600100761
-
[38]
W. C. Swope, H. C. Andersen, P. H. Berens, K. R. Wilson, A com- puter simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small wa- ter clusters, The Journal of Chemical Physics 76 (1982) 637–649.doi: 10.1063/1.442716
-
[39]
L. Berthier, W. Kob, The Monte Carlo dynamics of a binary Lennard-Jones glass-forming mixture, Journal of Physics: Condensed Matter 19 (2007) 205130.doi:10.1088/0953-8984/19/20/205130. 24
-
[40]
S. Sastry, P. G. Debenedetti, F. H. Stillinger, Signatures of distinct dynam- ical regimes in the energy landscape of a glass-forming liquid, Nature 393 (1998) 554–557.doi:10.1038/31189
-
[41]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Communi- c...
-
[42]
M. E. J. Newman, G. T. Barkema, Monte Carlo Methods in Sta- tistical Physics, Oxford University Press, 1999.doi:10.1093/oso/ 9780198517962.001.0001
-
[43]
V. Ramasubramani, B. D. Dice, E. S. Harper, M. P. Spellings, J. A. Ander- son, S. C. Glotzer, freud: A Software Suite for High Throughput Analysis of Particle Simulation Data, Computer Physics Communications 254 (2020) 107275.doi:10.1016/j.cpc.2020.107275
-
[44]
H. Yoshida, Construction of higher order symplectic integrators, Physics Letters A 150 (1990) 262–268.doi:10.1016/0375-9601(90)90092-3
-
[45]
K. Shiraishi, E. Minamitani, K. Kim, Dataset: Benchmarking thermostat algorithms in molecular dynamics simulations of a binary lennard-jones glass-former model (2025).doi:10.5281/zenodo.17081934. 25
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.