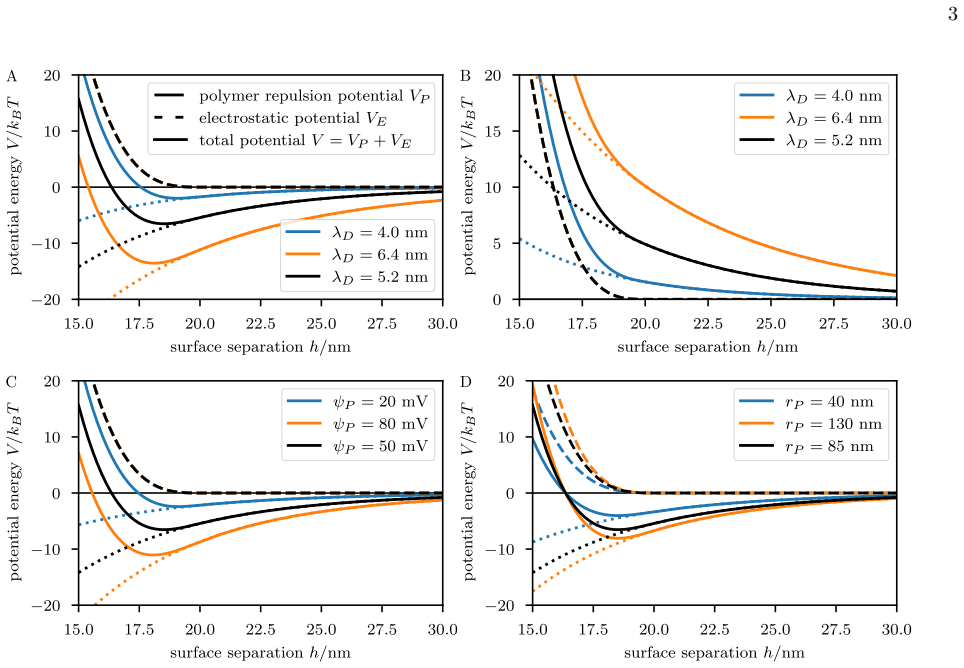
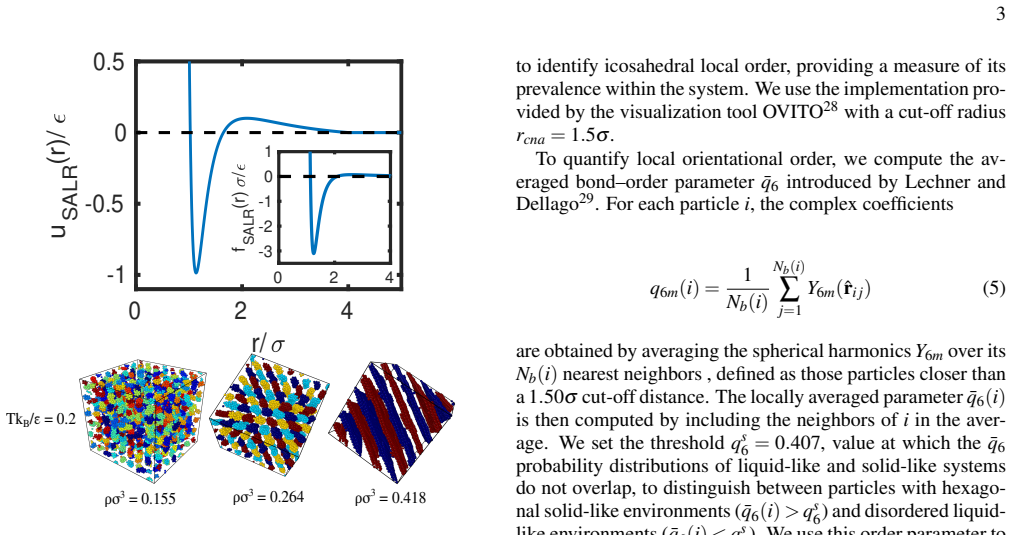
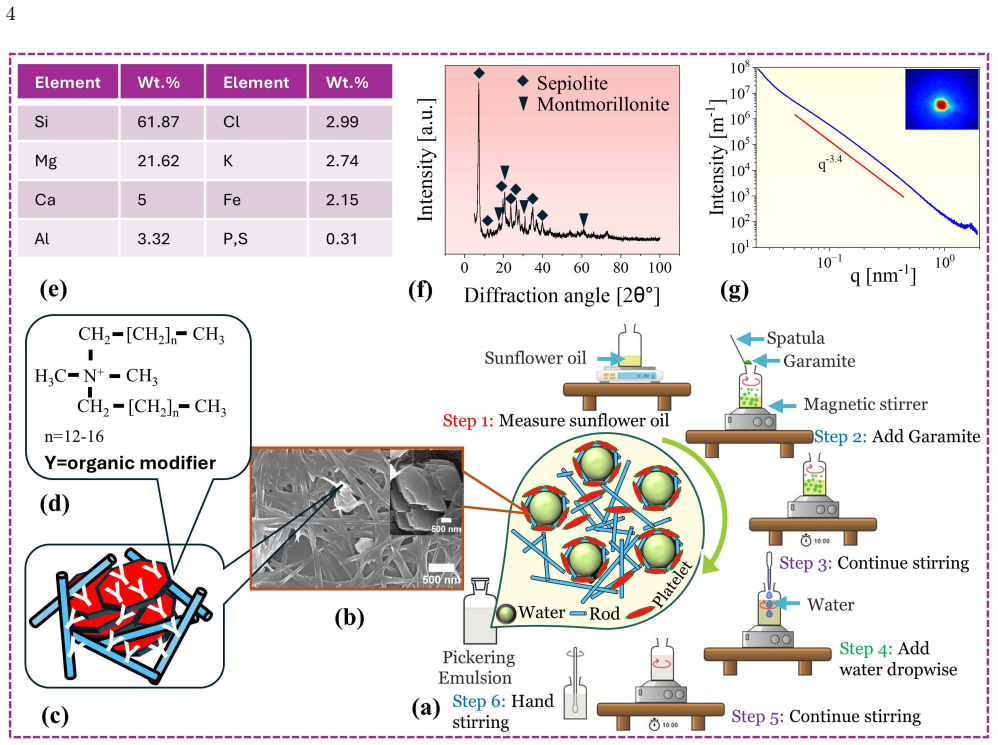
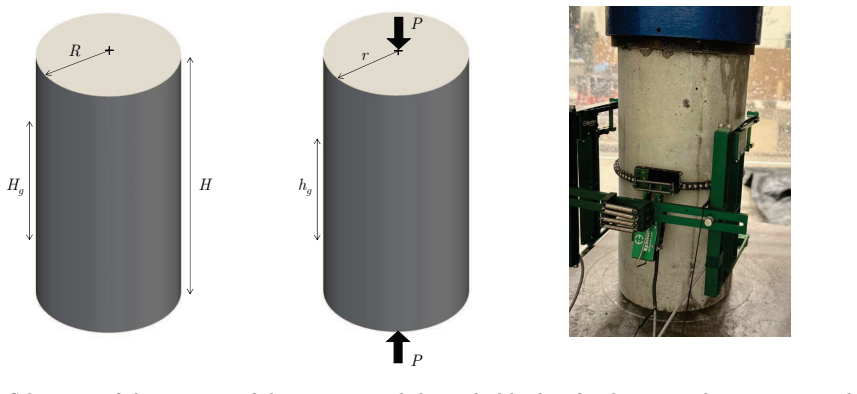
0
Active particles turn coffee rings into designed deposits
From Coffee Rings to Self-Driven Assembly: Active Matter Enabled Design of Drying Droplets
Motile colloids and microbes generate internal flows that replace passive capillary transport in drying droplets.
abstract click to expand
Evaporating colloidal droplets have long been used as model systems to understand capillarity, interfacial transport, and particle assembly, most prominently through the coffee ring effect. In classical descriptions, suspended particles are treated as passive tracers carried by evaporation-driven capillary flow, with additional influence from Marangoni stresses, wettability, and contact line pinning. More recent studies, however, show that this picture changes significantly when the particles themselves are active. Systems containing motile microorganisms, chemically active colloids, or externally driven particles can continuously inject energy or generate gradients within the droplet, leading to self-driven flows, modified interfacial stresses, and dynamic contact line behavior. In this Perspective, we bring together these developments, identify the key mechanisms governing active droplets, highlight the role of bubble-mediated flows, and outline strategies for controlled deposition and functional interface design.