Mechanistic Insights into Chemical Exchange during the Signal Amplification by Reversible Exchange Sensitization of Pyruvate
Pith reviewed 2026-05-16 17:03 UTC · model grok-4.3
The pith
Intramolecular hydride exchange outpaces pyruvate and H2 loss in SABRE sensitization
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Intramolecular hydrogen exchange of the hydrides occurs faster than pyruvate or H2 loss; a novel stable [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex is present; counterions such as Na+ influence Ir-pyruvate binding; and the populations of species shift with temperature, DMSO concentration, pyruvate concentration, and hydrogen pressure.
What carries the argument
Parahydrogen-enhanced spin-selective NMR combined with an exchange kinetic model and DFT structure calculations to track pyruvate binding and hydride movements on the iridium center.
If this is right
- Hydride intramolecular exchange is the fastest process and controls the overall SABRE kinetics for pyruvate.
- The iridium catalyst exists in a previously unrecognized stable form with one pyruvate and two DMSO ligands.
- Sodium counterions participate in modulating pyruvate attachment to the metal center.
- Species distributions and exchange rates change measurably with temperature, DMSO level, pyruvate level, and hydrogen pressure.
Where Pith is reading between the lines
- The same NMR approach could map exchange pathways for other substrates that SABRE has not yet polarized efficiently.
- Explicit inclusion of counterion effects in catalyst screening may raise achievable polarization levels.
- Measured exchange rates could be used to predict which solvent or salt additives will improve SABRE performance.
- The method supplies a general template for studying short-lived complexes in other parahydrogen-based polarization schemes.
Load-bearing premise
The kinetic model fitted to the NMR data includes every important species and pathway and the DFT calculations correctly identify the structures and relative energies of the observed complexes.
What would settle it
An NMR experiment that measures the hydride intramolecular exchange rate as equal to or slower than the rate of pyruvate dissociation, or that fails to detect the [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex under the stated conditions.
Figures
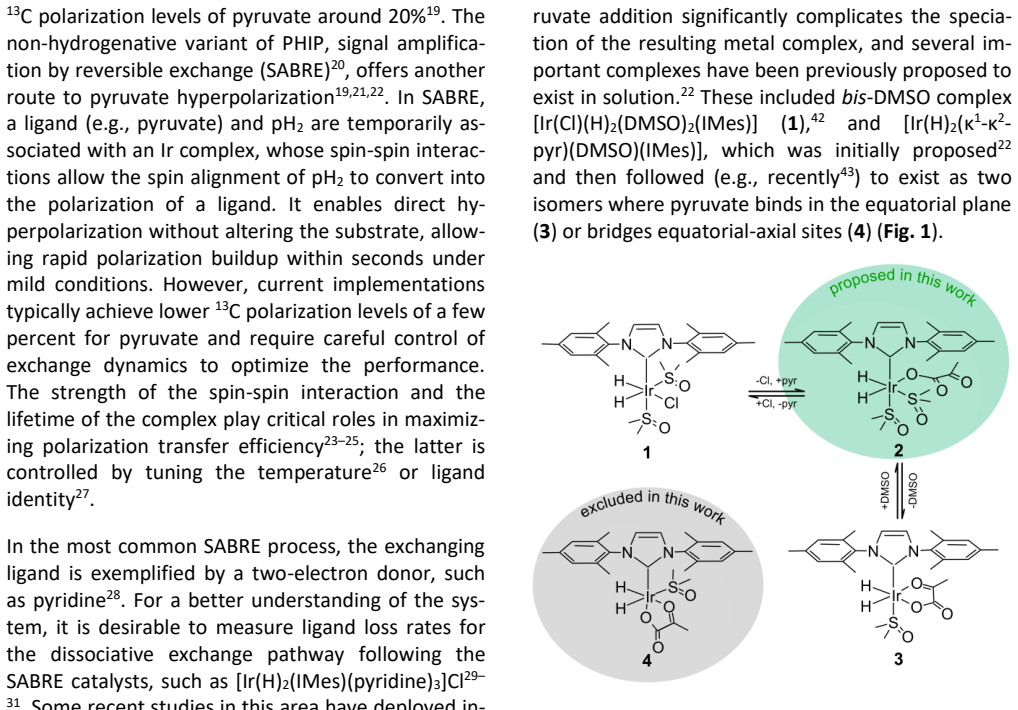
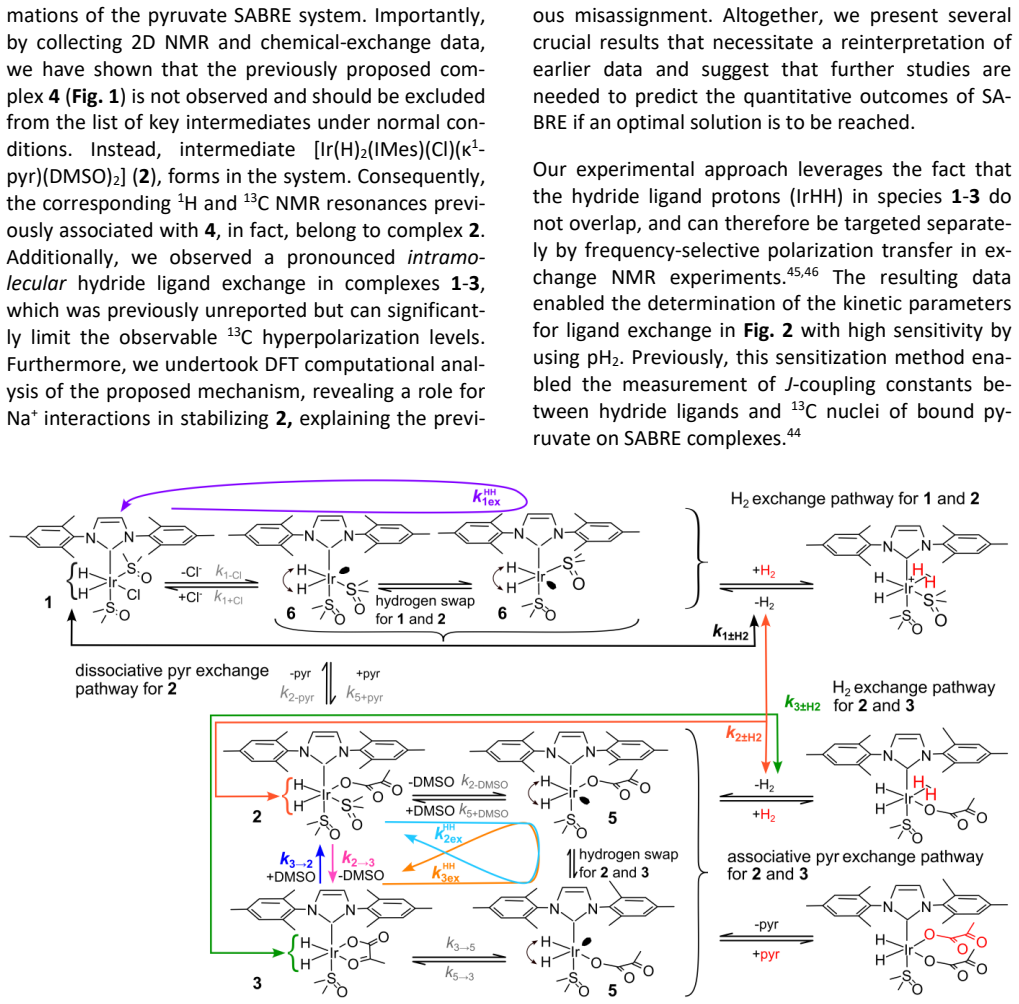
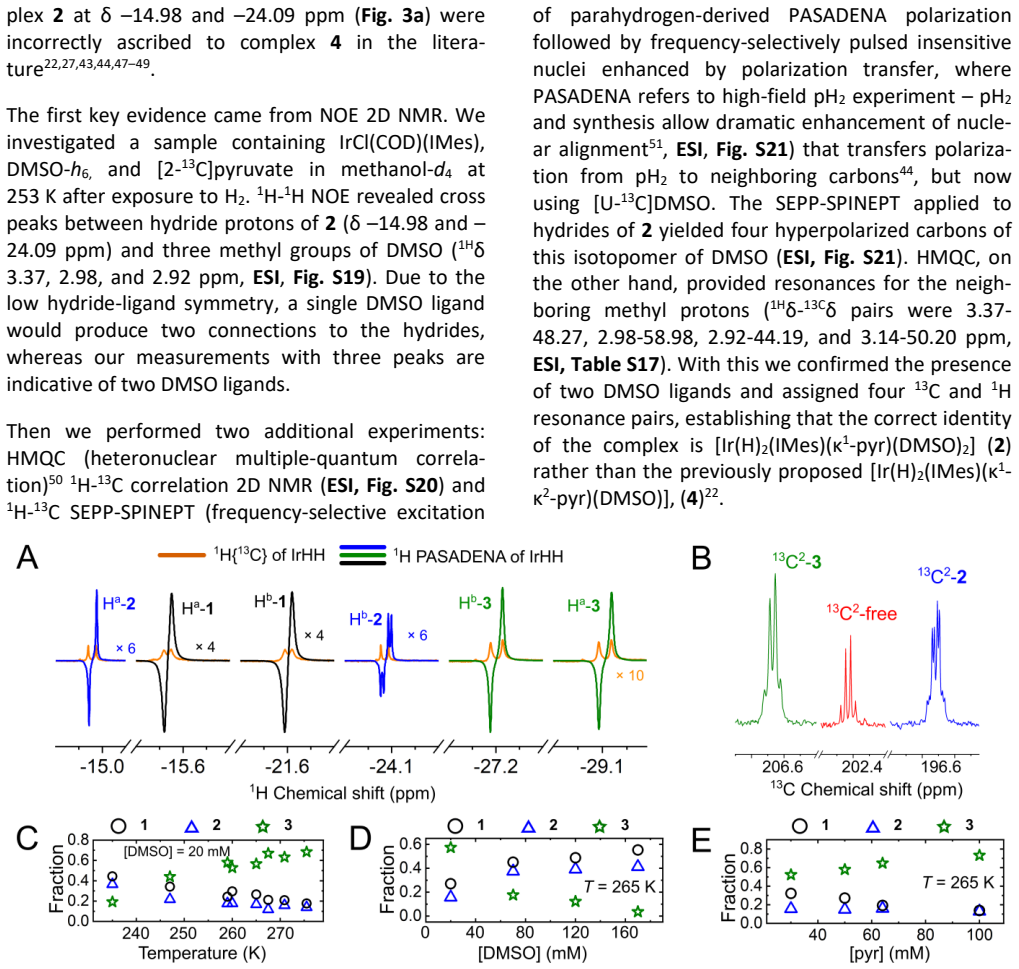
read the original abstract
Signal amplification by reversible exchange (SABRE) is a nuclear spin hyperpolarization technique in which the transient interaction of parahydrogen (pH2) and a target substrate with an iridium complex leads to polarization transfer to the substrate. Here, we use a parahydrogen-enhanced, spin-selective NMR method to investigate pyruvate binding, which is combined with exchange-model fitting and DFT calculations. Our study reveals several key findings that reshape the current understanding of SABRE: (a) intramolecular hydrogen exchange of the hydrides, occurring faster than pyruvate or H2 loss; (b) the discovery of a novel stable [Ir(H)2(IMes)(\k{appa}1-pyr)(DMSO)2] complex; and (c) the potential role of counterions (here Na+) in Ir-pyruvate binding. Previously unknown insights into complex kinetics and distributions as a function of temperature, [DMSO], [pyruvate], and hydrogen pressure are presented. The methods demonstrated here, exemplified by SABRE, provide a framework that is expected to guide future research in the field.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses parahydrogen-enhanced spin-selective NMR, multi-species exchange-model fitting, and DFT calculations to probe SABRE kinetics for pyruvate. It reports that intramolecular hydride exchange on the Ir complex is faster than pyruvate or H2 loss, identifies a previously unobserved stable [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] species, and proposes a role for Na+ counterions in modulating pyruvate binding, with additional data on temperature, [DMSO], [pyruvate], and H2-pressure dependence.
Significance. If the central rate ordering and complex identification hold, the work supplies concrete mechanistic constraints that can guide catalyst and solvent optimization in SABRE, and the combined NMR–fitting–DFT workflow offers a reusable template for other hyperpolarization systems.
major comments (3)
- [Section 3] Section 3 (kinetic modeling): the claim that intramolecular hydride exchange is faster than pyruvate/H2 loss rests on rates extracted from a single multi-species exchange model; no global fits across concentration series or isotopic-labeling controls are shown that would exclude omitted reversible pathways (e.g., DMSO-mediated or counterion-assisted routes) whose contribution could invert the ordering.
- [Abstract and Section 4] Abstract and Section 4 (results): fitted rate constants are presented without reported uncertainties, covariance matrices, or model-validation statistics (χ², residual analysis, or alternative-scheme comparisons), so the quantitative ordering cannot be assessed for robustness.
- [DFT section] DFT section (structure assignments): the novel [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex is assigned on the basis of computed energies and NMR parameters, yet no experimental cross-check (e.g., concentration-dependent speciation or 2D exchange spectra) is provided to confirm that this species is the dominant observed intermediate under the reported conditions.
minor comments (2)
- [Abstract] Abstract: the LaTeX fragment “κ1-pyr” is rendered as “k{appa}1-pyr”; correct the typesetting.
- [Methods and figures] Figure captions and methods: specify the exact temperature range, H2 pressure, and DMSO/pyruvate concentrations used for each data set to enable direct reproduction.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed review of our manuscript. We have addressed each major comment point-by-point below. Where the comments identify areas for strengthening the analysis, we have revised the manuscript accordingly and provide details on the changes.
read point-by-point responses
-
Referee: [Section 3] Section 3 (kinetic modeling): the claim that intramolecular hydride exchange is faster than pyruvate/H2 loss rests on rates extracted from a single multi-species exchange model; no global fits across concentration series or isotopic-labeling controls are shown that would exclude omitted reversible pathways (e.g., DMSO-mediated or counterion-assisted routes) whose contribution could invert the ordering.
Authors: We appreciate the referee's emphasis on rigorous validation of the kinetic model. The multi-species exchange model was constructed directly from the full set of observed NMR resonances and fitted simultaneously to the time-dependent polarization data collected across multiple experimental conditions (temperature, [DMSO], [pyruvate], and H2 pressure). The extracted rate ordering is consistent with these independent datasets. Nevertheless, we acknowledge that explicit global fitting across the full concentration series and additional discussion of alternative pathways would further strengthen the claim. In the revised manuscript we have added global-fit results and a dedicated paragraph addressing why DMSO- or counterion-mediated routes are unlikely to invert the observed ordering under the reported conditions. revision: yes
-
Referee: [Abstract and Section 4] Abstract and Section 4 (results): fitted rate constants are presented without reported uncertainties, covariance matrices, or model-validation statistics (χ², residual analysis, or alternative-scheme comparisons), so the quantitative ordering cannot be assessed for robustness.
Authors: We agree that uncertainties and model-validation metrics are necessary to evaluate the robustness of the quantitative rate ordering. In the revised manuscript we now report the standard errors obtained from the nonlinear least-squares fitting, the covariance matrix for the key rate constants, χ² values, residual plots, and explicit comparisons to two alternative kinetic schemes. These additions confirm that the intramolecular hydride-exchange rate remains the fastest process within the reported uncertainties. revision: yes
-
Referee: [DFT section] DFT section (structure assignments): the novel [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex is assigned on the basis of computed energies and NMR parameters, yet no experimental cross-check (e.g., concentration-dependent speciation or 2D exchange spectra) is provided to confirm that this species is the dominant observed intermediate under the reported conditions.
Authors: The assignment of the [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex is based on the close quantitative agreement between the experimental 1H and 13C chemical shifts and the DFT-computed values for this geometry, together with its persistence across the range of conditions examined. To provide the requested experimental cross-check, the revised manuscript now includes concentration-dependent speciation data and selected 2D EXSY spectra that confirm this species is the dominant observable intermediate under the conditions of the kinetic measurements. revision: yes
Circularity Check
No significant circularity; claims rest on spectroscopic detection and DFT rather than self-referential fits
full rationale
The paper extracts rate constants by fitting an exchange model to parahydrogen-enhanced NMR data and combines this with independent DFT calculations for complex structures. The central findings (intramolecular hydride exchange faster than loss processes, discovery of [Ir(H)2(IMes)(κ1-pyr)(DMSO)2], counterion role) are presented as direct observations from spectra and computations, not as predictions forced by the fit itself. No self-definitional equations, fitted inputs renamed as predictions, load-bearing self-citations, or ansatz smuggling appear in the abstract or described methods. The kinetic model serves as an interpretive tool whose assumptions are stated explicitly; the ordering claims remain data-driven and falsifiable against raw spectra. This yields a low circularity score consistent with normal use of fitting for mechanistic insight.
Axiom & Free-Parameter Ledger
free parameters (1)
- exchange rate constants
axioms (1)
- domain assumption Observed NMR signals arise exclusively from the proposed chemical species and exchange processes without significant overlap or unmodeled pathways.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
intramolecular hydrogen exchange of the hydrides, occurring faster than pyruvate or H2 loss; discovery of novel stable [Ir(H)2(IMes)(κ1-pyr)(DMSO)2] complex
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
kinetic exchange model used for fitting
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Nelson, S. J. et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1 - 13 C]Pyruvate. Sci. Transl. Med. 5, (2013)
work page 2013
-
[2]
Woitek, R. et al. Hyperpolarized 13 C MRI of Tumor Metabolism Demonstrates Early Metabolic Response to Neoadjuvant Chemotherapy in Breast Cancer. Ra- diol. Imaging Cancer 2, e200017 (2020)
work page 2020
-
[3]
Cunningham, C. H. et al. Hyperpolarized 13 C Metabol- ic MRI of the Human Heart: Initial Experience. Circ. Res. 119, 1177–1182 (2016)
work page 2016
-
[4]
Gallagher, F. A. et al. Imaging breast cancer using hyperpolarized carbon -13 MRI. Proc. Natl. Acad. Sci. 117, 2092–2098 (2020)
work page 2092
-
[5]
Granlund, K. L. et al. Hyperpolarized MRI of Human Prostate Cancer Reveals Increased Lactate with Tu- mor Grade Driven by Monocarboxylate Transporter 1. Cell Metab. 31, 105-114.e3 (2020)
work page 2020
-
[6]
Liberti, M. V. & Locasale, J. W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 41, 211–218 (2016)
work page 2016
-
[7]
Ardenkjær-Larsen, J. H. et al. Increase in signal -to- noise ratio of > 10,000 times in liquid -state NMR. Proc. Natl. Acad. Sci. 100, 10158–10163 (2003)
work page 2003
-
[8]
Eills, J. et al. Spin Hyperpolarization in Modern Mag- netic Resonance. Chem. Rev. 123, 1417–1551 (2023)
work page 2023
-
[9]
Hövener, J. et al. Parahydrogen‐Based Hyperpolariza‐ tion for Biomedicine. Angew. Chem. Int. Ed. 57, 11140–11162 (2018)
work page 2018
-
[10]
N., Buntkowsky, G., Duckett, S
Pravdivtsev, A. N., Buntkowsky, G., Duckett, S. B., Koptyug, I. V. & Hövener, J. Parahydrogen‐Induced Polarization of Amino Acids. Angew. Chem. Int. Ed. 60, 23496–23507 (2021)
work page 2021
- [11]
-
[12]
Brahms, A. et al. Exceptionally Mild and High -Yielding Synthesis of Vinyl Esters of Alpha -Ketocarboxylic Ac- ids, Including Vinyl Pyruvate, for Parahydrogen - Enhanced Metabolic Spectroscopy and Imaging. J. Org. Chem. 88, 15018–15028 (2023)
work page 2023
-
[13]
Ding, Y. et al. Rapidly Signal-enhanced Metabolites for Atomic Scale Monitoring of Living Cells with Magnetic Resonance. Chem Methods 2, e202200023 (2022)
work page 2022
-
[14]
Cavallari, E. et al. In-vitro NMR Studies of Prostate Tumor Cell Metabolism by Means of Hyperpolarized [1-13C]Pyruvate Obtained Using the PHIP -SAH Meth- od. Front. Oncol. 10, 497 (2020)
work page 2020
-
[15]
Brahms, A. et al. Synthesis of 13C and 2H Labeled Vinyl Pyruvate and Hyperpolarization of Pyruvate. Chem. – Eur. J. 28, e202201210 (2022)
work page 2022
-
[16]
Chukanov, N. V. et al. Synthesis of Unsaturated Pre- cursors for Parahydrogen -Induced Polarization and Molecular Imaging of 1 - 13 C-Acetates and 1 - 13 C- Pyruvates via Side Arm Hydrogenation. ACS Omega 3, 6673–6682 (2018)
work page 2018
-
[17]
Carrera, C. et al. ParaHydrogen Polarized Ethyl -[1- 13C]pyruvate in Water, a Key Substrate for Fostering the PHIP-SAH Approach to Metabolic Imaging. Chem- PhysChem 22, 1042–1048 (2021)
work page 2021
-
[18]
Dagys, L. et al. Nuclear hyperpolarization of (1 - 13 C)- pyruvate in aqueous solution by proton -relayed side- arm hydrogenation. The Analyst 146, 1772 –1778 (2021)
work page 2021
-
[19]
De Maissin, H. et al. In Vivo Metabolic Imaging of [1‐ 13 C]Pyruvate‐d 3 Hyperpolarized By Reversible Ex- change With Parahydrogen**. Angew. Chem. Int. Ed. 62, e202306654 (2023)
work page 2023
-
[20]
Adams, R. W. et al. Reversible Interactions with para - Hydrogen Enhance NMR Sensitivity by Polarization Transfer. Science 323, 1708–1711 (2009)
work page 2009
-
[21]
MacCulloch, K. et al. Facile hyperpolarization chemis- try for molecular imaging and metabolic tracking of [1–13C]pyruvate in vivo. J. Magn. Reson. Open 16–17, 100129 (2023)
work page 2023
-
[22]
Iali, W. et al. Hyperpolarising Pyruvate through Signal Amplification by Reversible Exchange (SABRE). An- gew. Chem. Int. Ed. 58, 10271–10275 (2019)
work page 2019
-
[23]
Barskiy, D. A., Pravdivtsev, A. N., Ivanov, K. L., Kovtu- nov, K. V. & Koptyug, I. V. A simple analytical model for signal amplification by reversible exchange (SA- BRE) process. Phys. Chem. Chem. Phys. 18, 89 –93 (2015)
work page 2015
-
[24]
Pravdivtsev, A. N. & Hövener, J. -B. Coherent polariza- tion transfer in chemically exchanging systems. Phys. Chem. Chem. Phys. 22, 8963–8972 (2020)
work page 2020
-
[25]
Van Weerdenburg, B. J. A. et al. Ligand effects of NHC–iridium catalysts for signal amplification by re- versible exchange (SABRE). Chem. Commun. 49, 7388 (2013)
work page 2013
-
[26]
TomHon, P. et al. Temperature Cycling Enables Effi- cient 13 C SABRE-SHEATH Hyperpolarization and Imag- ing of [1 - 13 C]-Pyruvate. J. Am. Chem. Soc. 144, 282– 287 (2022)
work page 2022
-
[27]
Tickner, Ben. J. et al. Optimisation of pyruvate hy- perpolarisation using SABRE by tuning the active magnetisation transfer catalyst. Catal. Sci. Technol. 10, 1343–1355 (2020)
work page 2020
-
[28]
Dücker, E. B., Kuhn, L. T., Münnemann, K. & Griesing- er, C. Similarity of SABRE field dependence in chemi- cally different substrates. J. Magn. Reson. 214, 159 – 165 (2012)
work page 2012
-
[29]
Lloyd, L. S. et al. Hyperpolarisation through reversible interactions with parahydrogen. Catal Sci Technol 4, 3544–3554 (2014)
work page 2014
-
[30]
Rayner, P. J. et al. Delivering strong 1 H nuclear hy- perpolarization levels and long magnetic lifetimes through signal amplification by reversible exchange. Proc. Natl. Acad. Sci. 114, (2017)
work page 2017
-
[31]
Rayner, P. J. et al. Fine-tuning the efficiency of para - hydrogen-induced hyperpolarization by rational N - heterocyclic carbene design. Nat. Commun. 9, 4251 (2018)
work page 2018
-
[32]
Cowley, M. J. et al. Iridium N -Heterocyclic Carbene Complexes as Efficient Catalysts for Magnetization Transfer from para -Hydrogen. J. Am. Chem. Soc. 133, 6134–6137 (2011)
work page 2011
-
[33]
Pravdivtsev, A. N., Yurkovskaya, A. V., Zimmermann, H., Vieth, H. -M. & Ivanov, K. L. Enhancing NMR of in- sensitive nuclei by transfer of SABRE spin hyperpolari- zation. Chem. Phys. Lett. 661, 77–82 (2016)
work page 2016
-
[34]
Vaneeckhaute, E., Tyburn, J., Kempf, J. G., Martens, J. A. & Breynaert, E. Reversible Parahydrogen Induced Hyperpolarization of 15 N in Unmodified Amino Acids Unraveled at High Magnetic Field. Adv. Sci. 10, 2207112 (2023)
work page 2023
-
[35]
Hermkens, N. K. J., Feiters, M. C., Rutjes, F. P. J. T., Wijmenga, S. S. & Tessari, M. High field hyperpolariza- tion-EXSY experiment for fast determination of disso- ciation rates in SABRE complexes. J. Magn. Reson. 276, 122–127 (2017)
work page 2017
-
[36]
Salnikov, O. G. et al. Modeling Ligand Exchange Kinet- ics in Iridium Complexes Catalyzing SABRE Nuclear Spin Hyperpolarization. Anal. Chem. acs.analchem.4c01374 (2024) doi:10.1021/acs.analchem.4c01374
-
[37]
Assaf, C. D. et al. Analysis of chemical exchange in iridium N -heterocyclic carbene complexes using het- eronuclear parahydrogen -enhanced NMR. Commun. Chem. 7, 286 (2024)
work page 2024
-
[38]
Peters, J. P., Assaf, C. D., Hövener, J. -B. & Pravdivtsev, A. N. Compact magnetic field cycling system with the range from nT to 9.4 T exemplified with 13C relaxa- tion dispersion and SABRE -SHEATH hyperpolarization. Preprint at https://doi.org/10.48550/ARXIV.2506.08711 (2025)
-
[39]
Pravdivtsev, A. N. et al. Chemical Exchange Reaction Effect on Polarization Transfer Efficiency in SLIC - SABRE. J. Phys. Chem. A 122, 9107–9114 (2018)
work page 2018
-
[40]
Konsewicz, K., Laczkó, G., Pápai, I. & Zhivonitko, V. V. Activation of H 2 using ansa -aminoboranes: solvent effects, dynamics, and spin hyperpolarization. Phys. Chem. Chem. Phys. 26, 3197–3207 (2024)
work page 2024
-
[41]
Barskiy, D. A., Pravdivtsev, A. N., Ivanov, K. L., Kovtu- nov, K. V. & Koptyug, I. V. A simple analytical model for signal amplification by reversible exchange (SA- BRE) process. Phys. Chem. Chem. Phys. 18, 89 –93 (2016)
work page 2016
-
[42]
Tickner, Ben. J. & Duckett, S. B. Iridium trihydride and tetrahydride complexes and their role in catalytic po- larisation transfer from para hydrogen to pyruvate. Chem. Sci. 16, 1396–1404 (2025)
work page 2025
-
[43]
Mamone, S. et al. (De)coding SABRE of [1 -13 C]pyruvate. Phys. Chem. Chem. Phys. 27, 22924 – 22936 (2025)
work page 2025
-
[44]
Assaf, C. D. et al. J Coupling Constants of <1 Hz Enable 13 C Hyperpolarization of Pyruvate via Reversible Ex- change of Parahydrogen. J. Phys. Chem. Lett. 1195– 1203 (2024) doi:10.1021/acs.jpclett.3c02980
-
[45]
Sengstschmid, H., Freeman, R., Barkemeyer, J. & Bar- gon, J. A New Excitation Sequence to Observe the PASADENA Effect. J. Magn. Reson. A 120, 249 –257 (1996)
work page 1996
-
[46]
Pravdivtsev, A. N., Hövener, J. -B. & Schmidt, A. B. Frequency-Selective Manipulations of Spins allow Ef- fective and Robust Transfer of Spin Order from Para- hydrogen to Heteronuclei in Weakly -Coupled Spin Systems. ChemPhysChem 23, e202100721 (2022)
work page 2022
-
[47]
B., Zanoni, G., De Biasi, F., Rastrelli, F
Mascitti, B. B., Zanoni, G., De Biasi, F., Rastrelli, F. & Saielli, G. Predicting the NMR chemical shifts of hy- drides in SABRE -active Ir complexes by relativistic DFT. Phys. Chem. Chem. Phys. https://doi.org/10.1039/d5cp01214g (2025) doi:10.1039/d5cp01214g
-
[48]
Myers, J. Z. et al. Zero to ultralow magnetic field NMR of [ 1 − 13 C ] pyruvate and [ 2 − 13 C ] pyruvate ena‐ bled by SQUID sensors and hyperpolarization. Phys. Rev. B 109, 184443 (2024)
work page 2024
-
[49]
Adelabu, I. et al. Order-Unity 13C Nuclear Polarization of [1 -13C]Pyruvate in Seconds and the Interplay of Water and SABRE Enhancement. ChemPhysChem 23, e202100839 (2022)
work page 2022
-
[50]
Palmer, A. G., Cavanagh, J., Wright, P. E. & Rance, M. Sensitivity improvement in proton -detected two - dimensional heteronuclear correlation NMR spectros- copy. J. Magn. Reson. 1969 93, 151–170 (1991)
work page 1969
-
[51]
Bowers, C. R. & Weitekamp, D. P. Parahydrogen and synthesis allow dramatically enhanced nuclear align- ment. J. Am. Chem. Soc. 109, 5541–5542 (1987)
work page 1987
-
[52]
(Pergamon Press, Oxford ; New York, 1981)
Hydrogen and Deuterium . (Pergamon Press, Oxford ; New York, 1981)
work page 1981
-
[53]
McBride, S. J. et al. Scalable Hyperpolarized MRI Ena- bled by Ace‐SABRE of [1‐ 13 C]Pyruvate. Angew. Chem. Int. Ed. 64, e202501231 (2025)
work page 2025
-
[54]
Becke, A. D. A new mixing of Hartree –Fock and local density-functional theories. J. Chem. Phys. 98, 1372 – 1377 (1993)
work page 1993
-
[55]
Accurate Coulomb -fitting basis sets for H to Rn
Weigend, F. Accurate Coulomb -fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057 (2006)
work page 2006
-
[56]
Izsák, R. & Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 135, (2011)
work page 2011
-
[57]
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta va- lence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297 (2005)
work page 2005
- [58]
-
[59]
Garcia‐Ratés, M. & Neese, F. Effect of the Solute Cavi‐ ty on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 41, 922–939 (2020)
work page 2020
-
[60]
Garcia‐Ratés, M. & Neese, F. Efficient implementation of the analytical second derivatives of hartree –fock and hybrid DFT energies within the framework of the conductor‐like polarizable continuum model. J. Com- put. Chem. 40, 1816–1828 (2019)
work page 2019
-
[61]
Andrae, D., Häußermann, U., Dolg, M., Stoll, H. & Preuß, H. Energy -adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77, 123–141 (1990)
work page 1990
-
[62]
Caldeweyher, E., Bannwarth, C. & Grimme, S. Exten- sion of the D3 dispersion coefficient model. J. Chem. Phys. 147, 034112 (2017)
work page 2017
-
[63]
Software Update: The ORCA Program Sys- tem—Version 6.0
Neese, F. Software Update: The ORCA Program Sys- tem—Version 6.0. WIREs Comput. Mol. Sci. 15, (2025)
work page 2025
-
[64]
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012)
work page 2012
-
[65]
Tickner, B. J., Parker, R. R., Whitwood, A. C. & Duck- ett, S. B. Probing the Hydrogenation of Vinyl Sulfox- ides Using para -Hydrogen. Organometallics 38, 4377–4382 (2019)
work page 2019
-
[66]
Nasibulov, E. A. et al. Analysis of Nutation Patterns in Fourier-Transform NMR of Non -Thermally Polarized Multispin Systems. Z. Für Phys. Chem. 227, 929 –953 (2013)
work page 2013
-
[67]
Pravdivtsev, A. N. et al. LIGHT-SABRE Hyperpolarizes 1- 13 C-Pyruvate Continuously without Magnetic Field Cycling. J. Phys. Chem. C 127, 6744–6753 (2023)
work page 2023
-
[68]
Hövener, J. -B. et al. A continuous -flow, high - throughput, high -pressure parahydrogen converter for hyperpolarization in a clinical setting: A HIGH - THROUGHPUT PARAHYDROGEN CONVERTER FOR HY- PERPOLARIZATION. NMR Biomed. 26, 124 –131 (2013)
work page 2013
-
[69]
Pravdivtsev, A. N., Ellermann, F. & Hövener, J. -B. Se- lective excitation doubles the transfer of parahydro- gen-induced polarization to heteronuclei. Phys. Chem. Chem. Phys. 23, 14146–14150 (2021)
work page 2021
-
[70]
McConnell, H. M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 28, 430–431 (1958)
work page 1958
-
[71]
Schmidt, A. B. et al. Lifetime of Para hydrogen in Aqueous Solutions and Human Blood. ChemPhysChem 20, 2408–2412 (2019)
work page 2019
-
[72]
Pravdivtsev, A. N., Kozinenko, V. P. & Hövener, J. -B. Only Para -Hydrogen Spectroscopy (OPSY) Revisited: In-Phase Spectra for Chemical Analysis and Imaging. J. Phys. Chem. A 122, 8948–8956 (2018)
work page 2018
-
[73]
Kessler, H., Oschkinat, H., Griesinger, C. & Bermel, W. Transformation of homonuclear two -dimensional NMR techniques into one -dimensional techniques us- ing Gaussian pulses. J. Magn. Reson. 1969 70, 106 – 133 (1986)
work page 1969
-
[74]
Bauer, C., Freeman, R., Frenkiel, T., Keeler, J. & Shaka, A. J. Gaussian pulses. J. Magn. Reson. 1969 58, 442– 457 (1984)
work page 1969
-
[75]
Schmidt, A. B. et al. Over 20% Carbon -13 Polarization of Perdeuterated Pyruvate Using Reversible Exchange with Parahydrogen and Spin -Lock Induced Crossing at 50 μT. J. Phys. Chem. Lett. 14, 5305–5309 (2023)
work page 2023
-
[76]
Knecht, S., Pravdivtsev, A. N., Hövener, J. -B., Yurkovskaya, A. V. & Ivanov, K. L. Quantitative de- scription of the SABRE process: rigorous consideration of spin dynamics and chemical exchange. RSC Adv. 6, 24470–24477 (2016)
work page 2016
-
[77]
Supramolecular Binding Thermodynamics by Dispersion‐Corrected Density Functional Theory
Grimme, S. Supramolecular Binding Thermodynamics by Dispersion‐Corrected Density Functional Theory. Chem. – Eur. J. 18, 9955–9964 (2012)
work page 2012
-
[78]
Perdew, J. P. et al. Atoms, molecules, solids, and sur- faces: Applications of the generalized gradient ap- proximation for exchange and correlation. Phys. Rev. B 46, 6671–6687 (1992)
work page 1992
-
[79]
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
work page 1996
-
[80]
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional ap- proximations. J. Chem. Phys. 105, 9982–9985 (1996)
work page 1996
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.