Recognition: 2 theorem links
· Lean TheoremMolecular g-Tensors From Spin-Orbit Quasidegenerate N-electron Valence Perturbation Theory: Benchmarks, Intruder-State Mitigation, and Practical Guidelines
Pith reviewed 2026-05-15 20:20 UTC · model grok-4.3
The pith
SO-QDNEVPT2 computes accurate g-tensors by treating electron correlation and spin-orbit coupling together.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
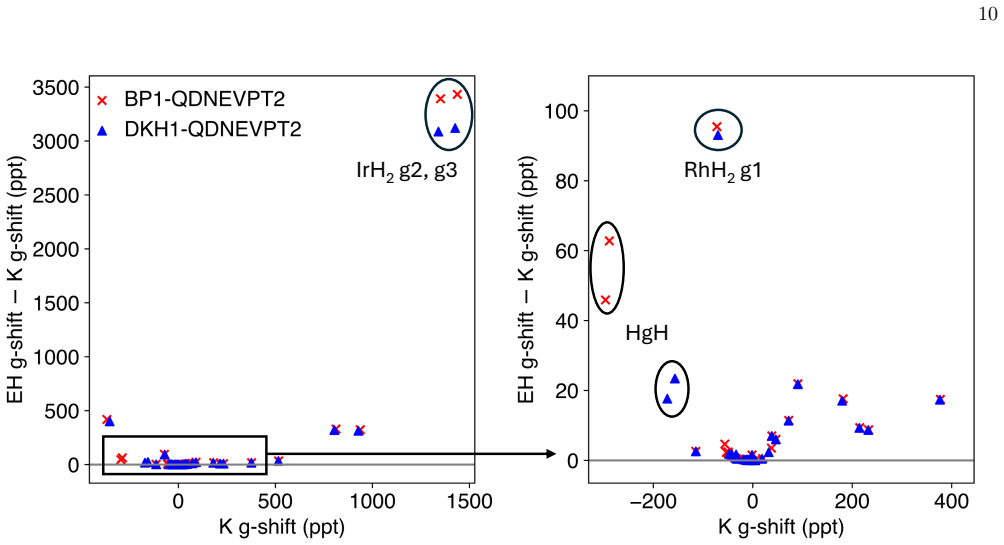
SO-QDNEVPT2 improves g-tensor predictions for correlated relativistic open-shell molecules by constructing a multistate effective Hamiltonian that incorporates both dynamical correlation and spin-orbit coupling. Benchmarks on 23 molecules demonstrate better agreement with experiment than SA-CASSCF. The Kramers approach is required for large g-shifts and intruder states are controlled by level-shift or renormalization techniques.
What carries the argument
The SO-QDNEVPT2 multistate effective Hamiltonian that folds second-order dynamical correlation and spin-orbit coupling into a single operator for g-tensor extraction.
If this is right
- SO-QDNEVPT2 yields closer agreement with experiment than state-averaged CASSCF on the 23-molecule benchmark.
- The Kramers approach from spin-mixed states is essential when g-shifts are large.
- Level-shift or renormalization techniques effectively remove intruder-state instabilities.
- Results vary with active space, state count, averaging weights, gauge origin and basis set, supplying practical guidelines.
Where Pith is reading between the lines
- The guidelines may help calculations on larger transition metal complexes beyond the current set.
- The intruder mitigation could extend to related properties like zero-field splitting.
- Further tests on systems with very strong spin-orbit coupling would check the framework's limits.
Load-bearing premise
The benchmark performance on 23 small molecules will generalize to larger or more strongly coupled systems without uncontrolled errors.
What would settle it
An experimental g-tensor for a larger open-shell molecule that shows a large deviation from the SO-QDNEVPT2 prediction when using the paper's recommended parameters and mitigation.
Figures














read the original abstract
Accurate prediction of molecular g-tensors for open-shell systems requires a balanced treatment of multireference electron correlation and relativistic spin-orbit coupling. Here, we develop and benchmark spin-orbit quasidegenerate second-order N-electron valence perturbation theory (SO-QDNEVPT2) for g-tensor calculations, treating dynamical correlation and spin-orbit effects consistently within a multistate effective Hamiltonian framework. Two g-tensor approaches are implemented: a spin-free effective Hamiltonian (EH) approach based on second-order response and a Kramers (K) approach that extracts g from spin-mixed SO-QDNEVPT2 states. We assess their performance on a benchmark set of 23 molecules spanning diatomics and small polyatomics, low- to high-spin species, and weak to strong spin-orbit coupling. Across the dataset, SO-QDNEVPT2 improves agreement with experiment relative to state-averaged complete active-space self-consistent field. The EH and K formalisms agree for modest g-shifts but the Kramers approach becomes essential when the shifts become large. We demonstrate that QDNEVPT2 results can be sensitive to intruder-state instabilities that can be effectively mitigated with level-shift or renormalization techniques. We then analyze the dependence of SO-QDNEVPT2 results on key computational parameters, including active space, number of states, state-averaging weights, gauge origin, and basis set. These results establish SO-QDNEVPT2 as a robust framework for computing g-tensors in correlated, relativistic open-shell molecules, offering practical guidelines for its applications.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops and benchmarks spin-orbit quasidegenerate second-order N-electron valence perturbation theory (SO-QDNEVPT2) for molecular g-tensor calculations in open-shell systems. It implements two approaches—an effective Hamiltonian (EH) method based on second-order response and a Kramers (K) method extracting g from spin-mixed states—within a multistate framework that treats dynamical correlation and spin-orbit coupling consistently. Performance is assessed on a 23-molecule benchmark set of diatomics and small polyatomics spanning low- to high-spin cases and weak to strong spin-orbit coupling, showing improvement over state-averaged CASSCF, with analysis of intruder-state mitigation via level shifts or renormalization and dependence on parameters such as active space, number of states, and basis set.
Significance. If the central claims hold, the work provides a useful correlated relativistic method for g-tensor prediction that improves upon SA-CASSCF while supplying concrete practical guidelines for parameter selection and intruder mitigation. This could support more reliable computational modeling of EPR spectra in open-shell molecules, particularly where multireference character and spin-orbit effects are important.
major comments (1)
- [Abstract and Benchmark Results] Benchmark description (abstract and results section): The robustness claim for SO-QDNEVPT2 as a framework rests on performance across the 23-molecule set of small systems. No additional benchmarks are shown for larger molecules or regimes with higher intruder-state density and stronger spin-orbit mixing, so it is unclear whether the reported level-shift/renormalization parameters and EH/K formalisms continue to control errors without degradation.
minor comments (1)
- [Abstract] The abstract states that results are sensitive to intruder states but does not quantify the magnitude of shifts before and after mitigation for the full set.
Simulated Author's Rebuttal
We thank the referee for the careful review and constructive comments on our manuscript. We address the major comment point by point below.
read point-by-point responses
-
Referee: Benchmark description (abstract and results section): The robustness claim for SO-QDNEVPT2 as a framework rests on performance across the 23-molecule set of small systems. No additional benchmarks are shown for larger molecules or regimes with higher intruder-state density and stronger spin-orbit mixing, so it is unclear whether the reported level-shift/renormalization parameters and EH/K formalisms continue to control errors without degradation.
Authors: We agree that the benchmark set is limited to 23 small molecules (diatomics and small polyatomics), selected to permit direct experimental comparison and systematic analysis of active-space, state-averaging, and intruder-state parameters. Larger systems with denser intruder manifolds or stronger SOC mixing are computationally more demanding at the CASSCF reference level and were outside the scope of the present validation. The level-shift and renormalization procedures we employ are general extensions of established QDNEVPT2 stabilization techniques that have been shown to remain effective in larger non-relativistic applications. We will add a paragraph in the Conclusions section explicitly stating the current scope, noting the expected transferability of the reported guidelines, and indicating that benchmarks on larger molecules are planned for future work. revision: partial
Circularity Check
No significant circularity: benchmarks compare to external experiment without fitting or self-definition
full rationale
The paper implements SO-QDNEVPT2 within a multistate effective Hamiltonian, computes g-tensors via EH and K formalisms, and benchmarks directly against experimental values on 23 molecules. No equations reduce computed g-tensors to parameters fitted from the target data; intruder-state mitigation and parameter dependence are analyzed as methodological choices, not self-referential predictions. The derivation chain remains self-contained against external benchmarks, with no load-bearing self-citations or ansatz smuggling that collapses the result to its inputs.
Axiom & Free-Parameter Ledger
free parameters (2)
- active space size
- number of states and averaging weights
axioms (1)
- domain assumption Multistate effective Hamiltonian framework consistently treats dynamical correlation and spin-orbit coupling
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
SO-QDNEVPT2 effective Hamiltonian ... intruder-state mitigation with level-shift or renormalization techniques
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
benchmark set of 23 molecules ... practical guidelines
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Effective Hamiltonian (EH) approach. The EH ap- proach follows the second-order treatment of spin–orbit and Zeeman interactions introduced by McWeeny, 98 in which both effects are included perturbatively within a scalar-relativistic, spin-free basis. Let |ΨSM 0 ⟩ denote a component ( M) of the spin multiplet of interest with total spin S, obtained as an e...
-
[2]
quasi-degenerate perturbation theory (QDPT)
Kramers (K) approach. To enable g-tensor calcu- lations using wavefunctions that already include spin– orbit coupling, we also employ a second strategy that starts from the spin-mixed SO-QDNEVPT2 eigenstates. In this approach, the Zeeman response of the spin- mixed manifold is mapped directly onto the effective spin Hamiltonian by equating the operators i...
-
[3]
Method abbreviation. Throughout this work, we denote each computational protocol for calculating g- tensors by A-B-C, where A specifies the spin–orbit treat- ment (BP1, BP2, DKH1, or DKH2), B specifies the electronic structure level (QDNEVPT2 or CASSCF), and C specifies the g-tensor evaluation strategy (K or EH). For example, DKH2-QDNEVPT2-K denotes calcu...
-
[4]
Active space size and number of states We first assess the sensitivity of ∆ g to (i) the defi- nition of the active space and (ii) the number of states included in the QDNEVPT2 effective Hamiltonian. Rep- resentative sets include p-shell diatomics with S = 1 /2 (ZnH, CdH, HgH; ZnF, CdF, HgF; CaH, SrH, BaH), d-shell transition metal hydrides (RhH 2, IrH 2,...
-
[5]
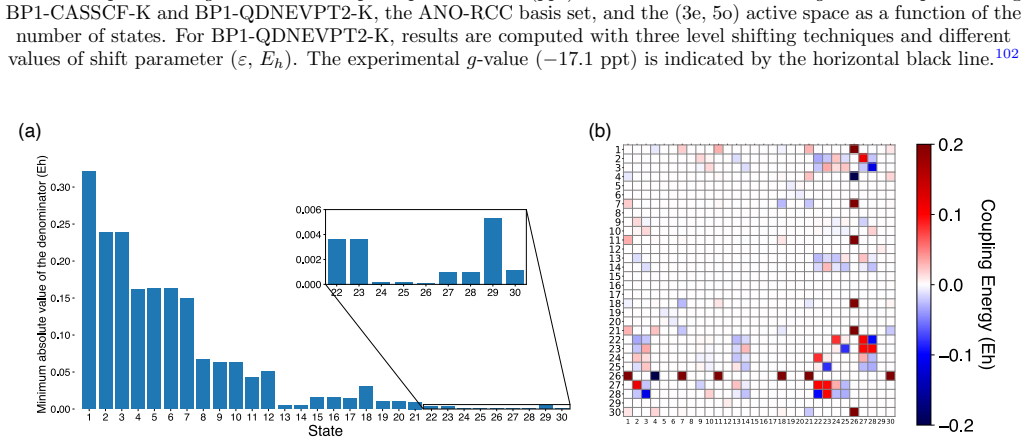
ZnH, CdH, and HgH. Figure 7a reports ∆ g⊥ for ZnH, CdH, and HgH computed using a small (3e, 5o) active space that incorporates the frontier s-orbitals of both atoms and three p-orbitals of the metal atom. For each molecule, the g-shifts are plotted as a function of the number of states included in the SO-QDNEVPT2 Hamiltonian, up to the maximum (50) permit...
-
[6]
CaH, SrH, and BaH. For these molecules, we exam- ined three active spaces: (i) (3e, 5o), comprising three σ and two π orbitals with dominant contributions from the atomic s-functions on both centers and from the metal p- and dz2-functions; (ii) (3e, 7o), obtained by adding two low-lying virtual δd-orbitals; and (iii) (3e, 9o), which further includes two v...
-
[7]
ZnF, CdF, HgF. Next, we investigate the results for the Zn–Hg fluoride series (Figure 9) that were com- puted using the (7e, 10o) active space incorporating the occupied σ-bond, two F p-lone pairs, the singly occupied σ-antibonding orbital, as well as two σ and two π vir- tual orbitals. As for the hydrides, the g-shifts originate from the p-shell configur...
-
[8]
RhH 2, IrH 2, and PdH. We next analyze the tran- sition metal hydrides RhH 2, IrH2, and PdH (Figures 10 to 12) with d-block elements near the middle of tran- sition metal series. The calculations employ (11e, 8o) active spaces for RhH 2 and IrH2 and (11e, 10o) for PdH (see the Supplementary Material). In this subset, the dominant spin–orbit response is co...
-
[9]
NCl, NBr, and NI. Finally, we consider the diatomic triplet radicals NCl, NBr, and NI, for which ∆ g⊥ was computed using the (12e, 8o) active space (Figure 13). As observed for other p-shell molecules in Section IV A, the BP–DKH separation grows with increasing nuclear charge (NCl < NBr < NI). Using fewer than ∼10 states leads to substantial errors in ∆ g...
-
[10]
State-averaging weights We next investigate the sensitivity of SO-QDNEVPT2 g-values to the SA-CASSCF reference weights by focus- ing on ZnH, CdH, and HgH (Figure 14) and the (3e, 5o) active space. Relative to equal-weight averaging (Fig- ure 14a), fixing 50% of the weight on the 2Σ ground state and distributing the residual weight among the excited states...
-
[11]
Gauge-origin error With finite basis sets, computed g-tensors can retain a residual dependence on the coordinate system origin. To quantify this effect, we shift the origin along the molec- ular axis for XH and XF (X = Zn, Cd, Hg), holding the metal atom at position 0, and compute the ∆ g⊥-values using the ANO-RCC basis set (Figure 15). We employ the (3e,...
-
[12]
Basis set effects We finally assess the basis set dependence of SO- QDNEVPT2 g-factors. Figure 16 compares ZnH, CdH, and HgH results obtained with the (3e, 5o) active space using the full ANO-RCC basis and the truncated ANO- RCC-VXZP (X = D, T, Q) sets. We use the ANO-RCC calculations as a reference to quantify errors introduced by basis set truncation in...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.