Assessing the impact of nodal surface optimization in fixed-node diffusion Monte Carlo on non-covalent interactions
Pith reviewed 2026-05-10 20:15 UTC · model grok-4.3
The pith
Optimizing the nodal surface in fixed-node diffusion Monte Carlo brings results closer to CCSD(T) for hydrogen-bonded non-covalent interactions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
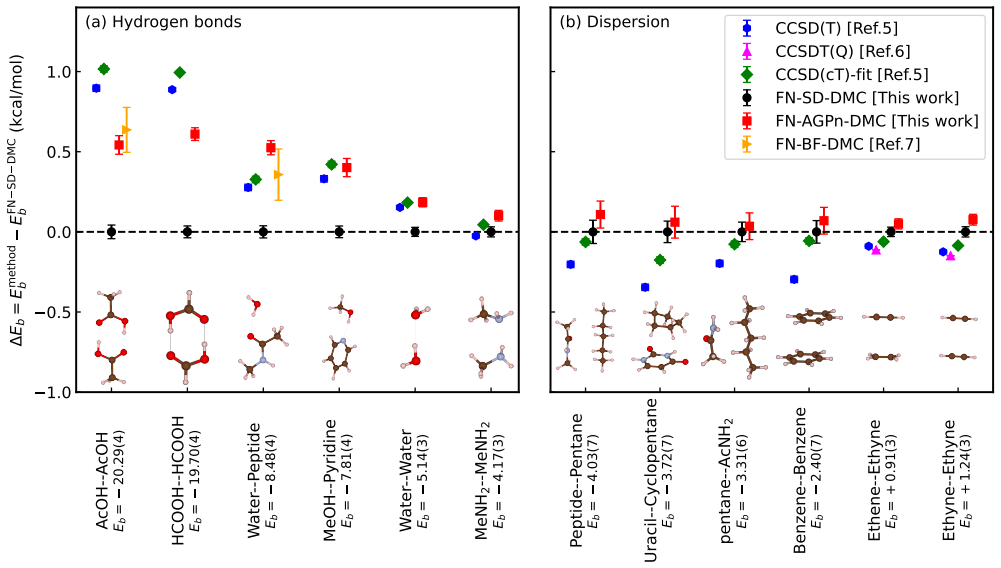
Adopting an antisymmetrized geminal power ansatz with natural orbitals to optimize the nodal surface in fixed-node diffusion Monte Carlo produces improved agreement with CCSD(T) reference values for hydrogen-bonded systems, while the same optimization has negligible effect on dispersion-dominated systems.
What carries the argument
Antisymmetrized geminal power (AGP) ansatz with natural orbitals, used to generate an improved nodal surface for fixed-node diffusion Monte Carlo.
If this is right
- Fixed-node DMC can achieve closer agreement with CCSD(T) for hydrogen-bonded non-covalent interactions by using the AGP nodal surface.
- The approach supplies a computationally modest route to reducing discrepancies specifically in hydrogen-bonded systems.
- Remaining differences between DMC and CCSD(T) in dispersion-dominated systems are unlikely to be removed by further nodal optimization alone.
- The pattern of improvement versus non-improvement distinguishes the dominant error source in the two classes of interactions.
Where Pith is reading between the lines
- Nodal quality appears more limiting for hydrogen-bonded complexes than for dispersion-bound ones, suggesting electron correlation near the nodes is stronger in the former.
- The same AGP nodal construction could be applied to larger clusters where CCSD(T) becomes prohibitive, providing an independent check on interaction energies.
- Dispersion systems may require improvements in the trial wavefunction beyond the nodes, such as better treatment of long-range correlation or finite-size effects.
Load-bearing premise
The AGP nodal surface lies close enough to the true nodes that any remaining fixed-node bias is not masked by compensating errors introduced by the optimization itself.
What would settle it
Repeating the calculations on the same hydrogen-bonded test set with an independent nodal-surface method that differs from both the original Slater determinant and the AGP surface and finding no systematic improvement over the original DMC results would falsify the central claim.
Figures

read the original abstract
Diffusion quantum Monte Carlo (DMC) and coupled cluster theory [CCSD(T)] are widely-employed benchmark methods for noncovalent interactions (NCIs). However, recent studies have reported notable discrepancies across several hydrogen-bonded and dispersion-dominated systems, raising questions on the accuracy of the approximations underlying each approach. In DMC, the dominant error is expected to stem from the fixed-node approximation, where the nodal surface is typically taken from a single Slater determinant derived from a density functional theory or Hartree-Fock calculation. In this work, we assess the impact of nodal surface optimization on DMC predictions for 12 compounds spanning diverse NCIs, using a recently proposed antisymmetrized geminal power ansatz with natural orbitals. We find improved agreement with CCSD(T) for hydrogen-bonded systems, while having negligible effect for dispersion-dominated systems. These results provide a practical and computationally efficient route to resolving discrepancies in hydrogen-bonded interactions, while offering insight into the remaining differences in dispersion-dominated systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript evaluates the effect of nodal surface optimization in fixed-node diffusion Monte Carlo using an antisymmetrized geminal power (AGP) ansatz constructed from natural orbitals. For a test set of 12 compounds spanning hydrogen-bonded and dispersion-dominated non-covalent interactions, the optimized nodes yield improved agreement with CCSD(T) reference values for the hydrogen-bonded subset while producing negligible changes for dispersion-dominated cases.
Significance. If the observed improvements reflect genuine reduction of fixed-node error rather than compensatory cancellation, the approach supplies a computationally tractable route to higher-accuracy DMC energies for hydrogen-bonded NCIs and clarifies the origin of remaining DMC–CCSD(T) discrepancies in dispersion systems. The work directly addresses a known source of benchmark inconsistency in the field.
major comments (1)
- [Abstract and Results] The central claim that AGP nodal optimization reduces fixed-node error (Abstract; Results section) rests on the untested premise that the AGP surface lies closer to the exact nodes. Because both DMC and CCSD(T) are approximate, the reported reduction in discrepancy for hydrogen-bonded systems could arise from a new AGP bias whose sign aligns with residual CCSD(T) error. No independent diagnostic—such as pure variational Monte Carlo energies, nodal overlap integrals, or comparison against a higher-level nodal reference—is presented to separate genuine node improvement from error cancellation.
Simulated Author's Rebuttal
We thank the referee for the careful review and for recognizing the potential significance of the work. The major comment is addressed below with proposed revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract and Results] The central claim that AGP nodal optimization reduces fixed-node error (Abstract; Results section) rests on the untested premise that the AGP surface lies closer to the exact nodes. Because both DMC and CCSD(T) are approximate, the reported reduction in discrepancy for hydrogen-bonded systems could arise from a new AGP bias whose sign aligns with residual CCSD(T) error. No independent diagnostic—such as pure variational Monte Carlo energies, nodal overlap integrals, or comparison against a higher-level nodal reference—is presented to separate genuine node improvement from error cancellation.
Authors: We agree that the manuscript does not present independent diagnostics (such as VMC energies or nodal overlaps) that would directly confirm the AGP nodes are closer to the exact nodes. Our primary finding is the observed improvement in agreement with CCSD(T) for the hydrogen-bonded subset and the lack of change for dispersion-dominated cases. We acknowledge that this improvement could in principle arise from a compensatory bias rather than genuine nodal improvement. To address this, we will revise the abstract and results section to phrase the conclusions strictly in terms of the change in DMC–CCSD(T) discrepancy rather than claiming a reduction in fixed-node error. We will also add a concise discussion paragraph noting the limitation and the value of future diagnostics. The system-dependent pattern (improvement only for hydrogen-bonded systems) is consistent with the physical expectation that nodal quality matters more when correlation is dominated by short-range effects, but we recognize this does not constitute proof. revision: yes
Circularity Check
No significant circularity; computational assessment relies on external benchmarks
full rationale
The manuscript conducts a numerical assessment of AGP-based nodal optimization in fixed-node DMC by direct comparison of computed interaction energies against independent CCSD(T) reference values for 12 systems. No load-bearing derivation, equation, or fitted parameter reduces the reported improvements (or lack thereof) to the input data by construction. The AGP ansatz is invoked as a recently proposed tool rather than derived within the paper, and the central claim rests on external falsifiable benchmarks rather than self-citation chains or self-definitional loops. This is the expected non-circular outcome for a benchmark-style computational study.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Fixed-node approximation error dominates DMC inaccuracies for these systems
- domain assumption Natural orbitals from a prior calculation provide a good starting point for AGP nodal optimization
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the dominant error is expected to stem from the fixed-node approximation, where the nodal surface is typically taken from a single Slater determinant derived from a density functional theory or Hartree-Fock calculation
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
FN-AGPn-DMC yields smaller binding energies than the FN-SD-DMC does; thus, provides bonding energies very close to those of CCSD(T)
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
X.; Della Pia, F.; Al-Hamdani, Y
Shi, B. X.; Della Pia, F.; Al-Hamdani, Y. S.; Michaelides, A.; Alf` e, D.; Zen, A. System- atic discrepancies between reference methods for noncovalent interactions within the S66 dataset.J. Chem. Phys.2025,162, 144107
work page 2025
-
[2]
Dubeck´ y, M. Noncovalent Interactions by Fixed-Node Diffusion Monte Carlo: Conver- gence of Nodes and Energy Differences vs Gaussian Basis-Set Size.J. Chem. Theory Comput.2017,13, 3626–3635
work page 2017
-
[3]
Nakano, K.; Shi, B. X.; Alf` e, D.; Zen, A. Basis Set Incompleteness Errors in Fixed- Node Diffusion Monte Carlo Calculations on Noncovalent Interactions.J. Chem. Theory Comput.2025,21, 4426–4434
work page 2025
-
[4]
Bennett, M. C.; Melton, C. A.; Annaberdiyev, A.; Wang, G.; Shulenburger, L.; Mitas, L. A new generation of effective core potentials for correlated calculations.J. Chem. Phys. 2017,147, 224106
work page 2017
-
[5]
G.; Michaelides, A.; Alf` e, D
Zen, A.; Brandenburg, J. G.; Michaelides, A.; Alf` e, D. A new scheme for fixed node diffusion quantum Monte Carlo with pseudopotentials: Improving reproducibility and reducing the trial-wave-function bias.J. Chem. Phys.2019,151, 134105
work page 2019
-
[6]
TurboRVB: A many-body toolkit for ab initio electronic simulations by quantum Monte Carlo.J
Nakano, K.; Attaccalite, C.; Barborini, M.; Capriotti, L.; Casula, M.; Coccia, E.; Dagrada, M.; Genovese, C.; Luo, Y.; Mazzola, G.; Zen, A.; Sorella, S. TurboRVB: A many-body toolkit for ab initio electronic simulations by quantum Monte Carlo.J. Chem. Phys.2020,152, 204121
work page 2020
-
[7]
Energy-consistent pseudopotentials for quantum Monte Carlo calculations.J
Burkatzki, M.; Filippi, C.; Dolg, M. Energy-consistent pseudopotentials for quantum Monte Carlo calculations.J. Chem. Phys.2007,126, 234105
work page 2007
-
[8]
Beyond the locality approximation in the standard diffusion Monte Carlo method.Phys
Casula, M. Beyond the locality approximation in the standard diffusion Monte Carlo method.Phys. Rev. B2006,74, 161102. s8
-
[9]
Wagner, L. K.; Bajdich, M.; Mitas, L. QWalk: A quantum Monte Carlo program for electronic structure.J. Comput. Phys.2009,228, 3390–3404
work page 2009
-
[10]
Trail, J. R.; Needs, R. J. Shape and energy consistent pseudopotentials for correlated electron systems.The Journal of Chemical Physics2017,146, 204107
-
[11]
Needs, R. J.; Towler, M. D.; Drummond, N. D.; L´ opez R´ ıos, P.; Trail, J. R. Variational and diffusion quantum Monte Carlo calculations with the CASINO code.J. Chem. Phys. 2020,152, 154106. s9
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.