The BOS-TMC Dataset: DFT Properties of 159k Experimentally Characterized Transition Metal Complexes Spanning Multiple Charge and Spin States
Pith reviewed 2026-05-10 16:53 UTC · model grok-4.3
The pith
The BOS-TMC dataset supplies DFT-computed properties for 159k experimentally characterized transition metal complexes across multiple charges and spin states.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
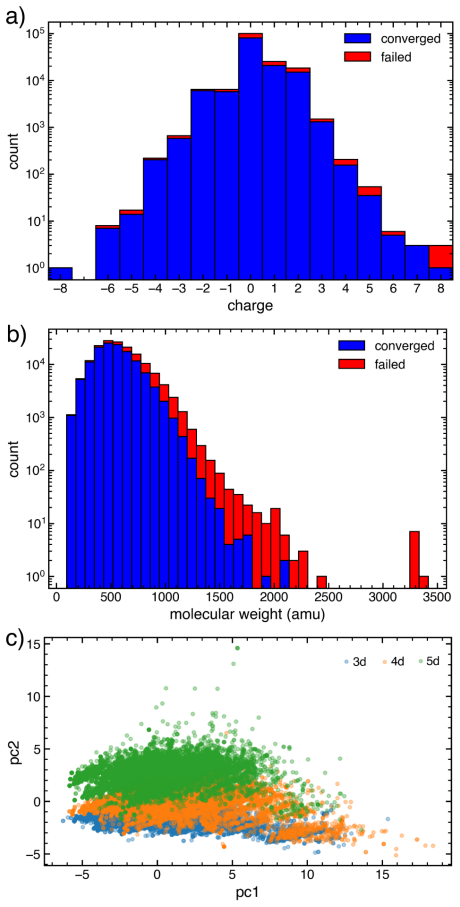
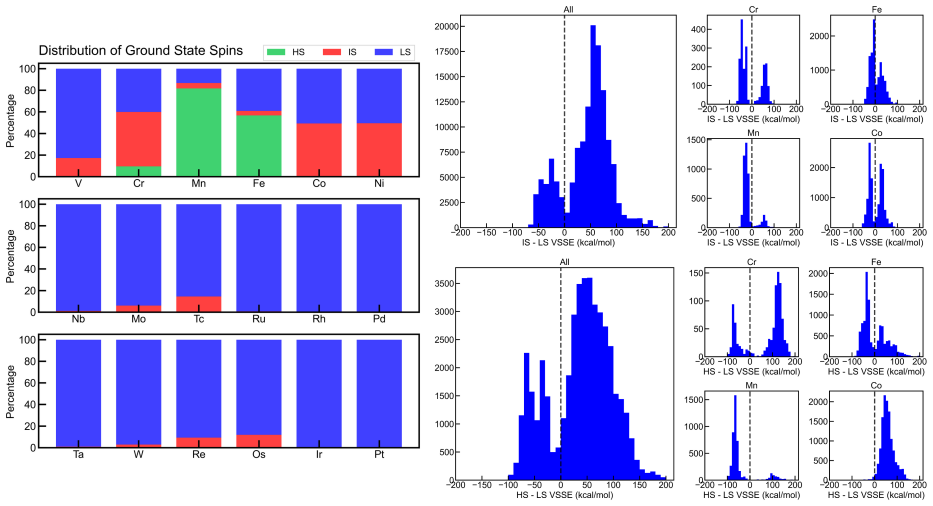
The paper introduces the Boston Open-Shell Transition Metal Complex dataset containing density functional theory properties for 159,000 mononuclear transition metal complexes in multiple spin states and formal charges, derived from the Cambridge Structural Database with experimental heavy-atom coordinates preserved during optimization and single-point energies evaluated at the PBE0/def2-TZVP level, along with a scheme for metal-spin-dependent atomization energies.
What carries the argument
The iterative procedure for confidently assigning overall TMC charge, combined with preservation of experimental heavy-atom coordinates during optimization and PBE0 single-point energy calculations on structures in compatible spin states.
Load-bearing premise
The iterative procedure confidently assigns overall TMC charge and that preserving experimental heavy-atom coordinates during optimization yields reliable properties across the diverse set of complexes and spin states.
What would settle it
Finding a substantial number of complexes where the assigned charges conflict with independently determined experimental charges or where fully relaxed geometries produce properties that differ enough from the fixed-coordinate results to change chemical conclusions.
Figures
read the original abstract
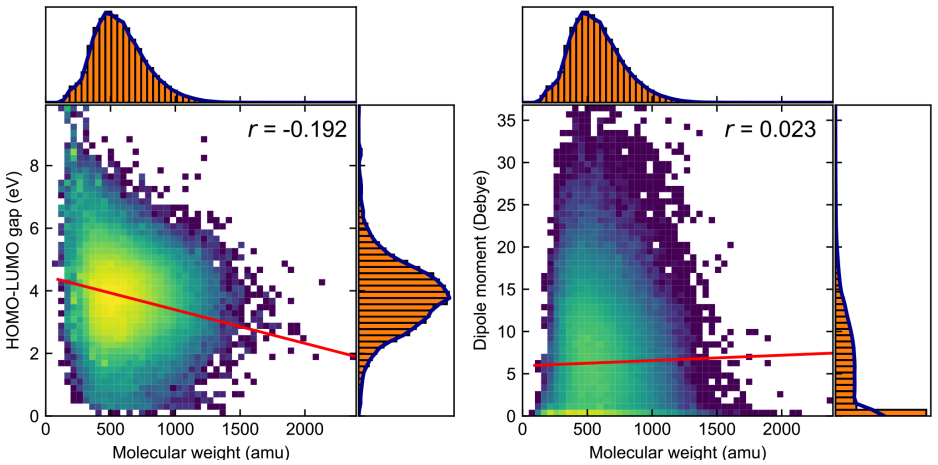
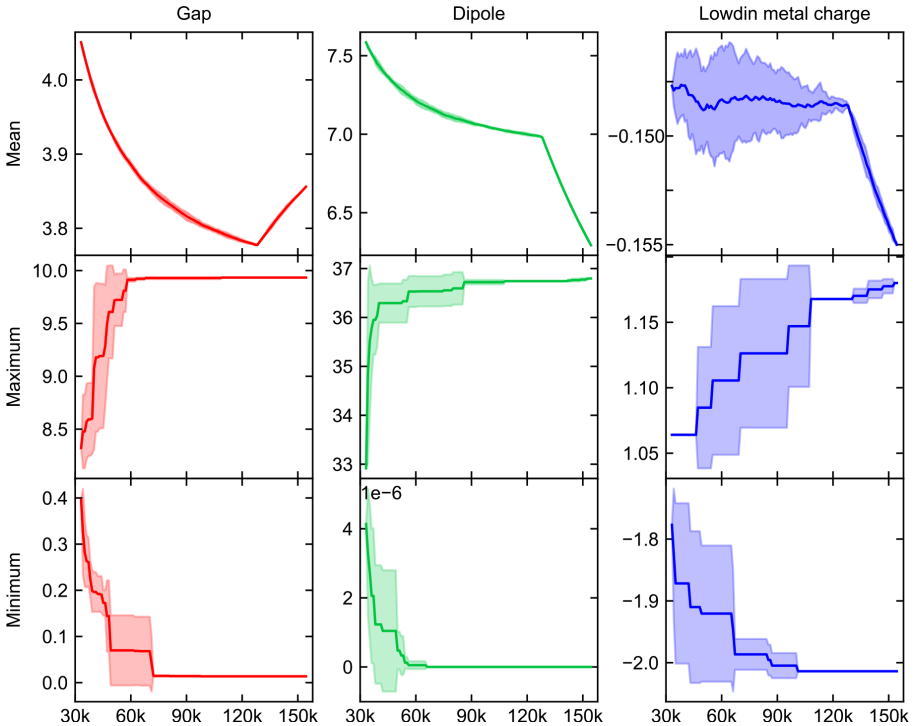
We present the Boston Open-Shell Transition Metal Complex (BOS-TMC) dataset, a set of density functional theory (DFT) properties for 159k experimentally characterized mononuclear transition metal complexes (TMCs) in multiple spin states with a range of formal charges derived from the Cambridge Structural Database (CSD). To curate this set, we carried out an iterative procedure to confidently assign overall TMC charge. From this information, we then obtained properties in up to three spin states, i.e., low-, intermediate-, and high-spin for 3d metals and low- and intermediate-spin for 4d and 5d metals, depending on compatibility with the metal electron configuration, for a total of 343.8k TMC/spin combinations. At odds with prior sets, we preserved experimental heavy-atom coordinates in these structures during optimization. We report all properties using PBE0/def2-TZVP single-point energies on these structures. We introduce a scheme for computing metal-spin-dependent atomization energies, which we report for each TMC. Alongside electronic energies, we report up to seven additional properties including: HOMO, LUMO, HOMO-LUMO gap, atomic partial charges, dipole moments, atomization energies, and spin-splitting energies for a total of over 2.9M TMC-associated properties. For a representative subset of over 10k complexes chosen based on size, we evaluate the sensitivity of computed properties to exchange-correlation (xc) functional choice from a set of twelve xcs spanning rungs of "Jacob's ladder", highlighting hotspots of TMC space that have the greatest uncertainty. In comparison to prior transition-metal datasets, BOS-TMC is both larger and more diverse in terms of charge and spin configurations and, as a result, more diverse in its range of properties. This dataset is expected to provide a high-fidelity foundation for machine-learning model development, DFT benchmarking, and exploration.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents the BOS-TMC dataset of DFT properties for 159k experimentally characterized mononuclear transition metal complexes (TMCs) drawn from the CSD, spanning multiple formal charges and spin states (low-, intermediate-, and high-spin where compatible) for a total of 343.8k TMC/spin combinations. The workflow uses an iterative charge-assignment procedure, preserves experimental heavy-atom coordinates during optimization, performs PBE0/def2-TZVP single-point calculations, introduces metal-spin-dependent atomization energies, and reports additional properties (HOMO, LUMO, gaps, partial charges, dipoles, spin splittings) totaling over 2.9M entries. A 10k-subset sensitivity analysis to twelve xc functionals is included, with the dataset positioned as larger and more diverse than prior TMC collections for ML, benchmarking, and exploration.
Significance. If the curation steps prove reliable, the dataset would provide a substantial, experimentally anchored resource for open-shell transition-metal chemistry. Its scale, coverage of charge/spin diversity, metal-spin-dependent atomization energies, and explicit functional-sensitivity study on a representative subset address documented gaps in existing TMC data collections and would support improved ML model training and DFT benchmarking in this challenging domain.
major comments (2)
- [Methods (iterative charge assignment)] The iterative charge-assignment procedure is load-bearing for selecting valid spin states and generating the 343.8k TMC/spin entries, yet the manuscript reports no quantitative validation metrics (success rate, agreement with literature charges for benchmark subsets, or error rates stratified by metal or ligand class).
- [Computational details and results (geometry handling)] Preservation of experimental heavy-atom coordinates is presented as ensuring high fidelity, but no comparison of key properties (atomization energies, spin splittings, HOMO-LUMO gaps) between fixed-geometry single points and fully DFT-optimized structures is provided, leaving the reliability of the reported values for the full set unquantified.
minor comments (2)
- [Abstract] Clarify whether atomization energies are included in the 'up to seven additional properties' count or reported separately, and ensure consistent terminology between abstract and main text.
- [Introduction] Add explicit numerical comparisons (size, charge/spin coverage, property ranges) to the prior TMC datasets referenced in the introduction to substantiate the 'larger and more diverse' claim.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review of our manuscript on the BOS-TMC dataset. We address each major comment below, indicating planned revisions where appropriate.
read point-by-point responses
-
Referee: The iterative charge-assignment procedure is load-bearing for selecting valid spin states and generating the 343.8k TMC/spin entries, yet the manuscript reports no quantitative validation metrics (success rate, agreement with literature charges for benchmark subsets, or error rates stratified by metal or ligand class).
Authors: We agree that explicit quantitative validation of the iterative charge-assignment procedure would strengthen the manuscript. The procedure applies standard electron-counting rules with iterative refinement to ensure consistency, but we acknowledge the absence of reported success rates or stratified comparisons in the original text. In the revised version, we will add a new subsection to the Methods section reporting the overall success rate, agreement with a manually verified benchmark subset of 500 complexes drawn from the literature, and error rates stratified by metal and ligand class. This addition will directly quantify the reliability of the curation step. revision: yes
-
Referee: Preservation of experimental heavy-atom coordinates is presented as ensuring high fidelity, but no comparison of key properties (atomization energies, spin splittings, HOMO-LUMO gaps) between fixed-geometry single points and fully DFT-optimized structures is provided, leaving the reliability of the reported values for the full set unquantified.
Authors: We thank the referee for this point. Preserving experimental heavy-atom coordinates was a deliberate choice to maintain fidelity to measured structures rather than allowing unconstrained optimization, which can introduce significant deviations in open-shell TMCs. To address the request for quantification, we will add an analysis in the revised manuscript comparing the specified properties (atomization energies, spin splittings, and HOMO-LUMO gaps) on a diverse representative subset of 5,000 complexes between the fixed-geometry single-point calculations and fully optimized structures. This will be reported in the Computational Details section. A full-set comparison is not feasible due to computational cost, but the subset will be selected to reflect the diversity of the dataset. revision: partial
Circularity Check
No circularity: standard data curation from external structures
full rationale
This is a dataset-generation paper that applies standard DFT (PBE0/def2-TZVP single-points) to experimentally determined CSD structures after an iterative charge-assignment curation step. No derivations, predictions, or first-principles results are claimed that reduce to fitted parameters or self-citations by construction. The charge-assignment procedure and fixed-geometry choice are methodological choices whose validity is external to any internal equation; they do not create a self-definitional loop or rename a fitted input as a prediction. All reported quantities (energies, gaps, atomization energies, etc.) are direct outputs of the chosen electronic-structure method on the curated inputs. No load-bearing self-citation chains or uniqueness theorems are invoked. The paper is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We present the Boston Open-Shell Transition Metal Complex (BOS-TMC) dataset, a set of density functional theory (DFT) properties for 159k experimentally characterized mononuclear transition metal complexes (TMCs) in multiple spin states with a range of formal charges derived from the Cambridge Structural Database (CSD).
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We introduce a scheme for computing metal-spin-dependent atomization energies, which we report for each TMC.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Calcs which completed but the graph changed: 1,026 a
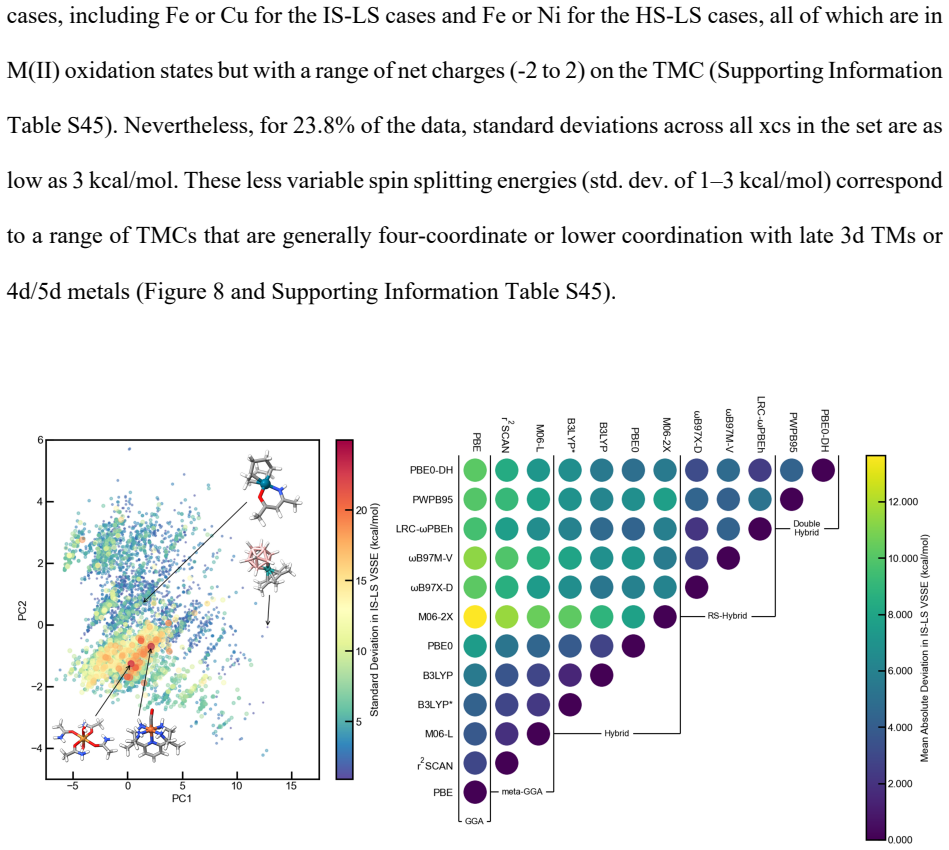
All attempted TeraChem calcs (total//unique): 162,109//128,357 2. Calcs which completed but the graph changed: 1,026 a. 22 of these were problematic (i.e., hydrogen atoms flew away from the complex, identified by any hydrogen atom being over 1.5 Å away from the nearest heavy atom) and marked as failures b. All others (1,004) were kept in (see explanation ...
-
[2]
In Psi4, attempt to converge the DFA with def2-TZVP starting from a functional that is not PBE0. a. In the case of semilocal DFAs, take a converged PBE result and initialize the calculations for other semilocal DFAs from PBE. i. If the PBE calculation did not converge, successively converge PBE+x% calculations, where x is the amount of Hartree-Fock exchan...
work page 2059
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.