PepBenchmark: A Standardized Benchmark for Peptide Machine Learning
Pith reviewed 2026-05-10 15:47 UTC · model grok-4.3
The pith
PepBenchmark supplies the first standardized benchmark for peptide machine learning through unified datasets, preprocessing, and evaluation protocols.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
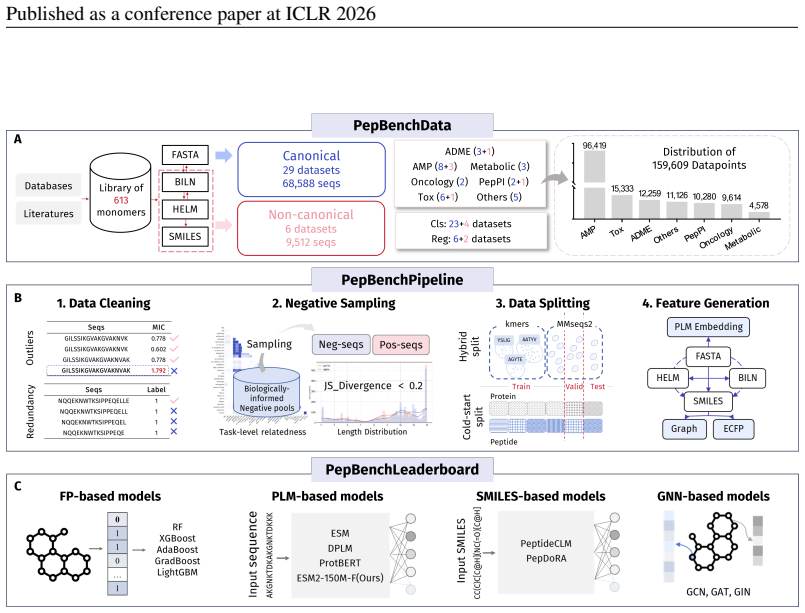
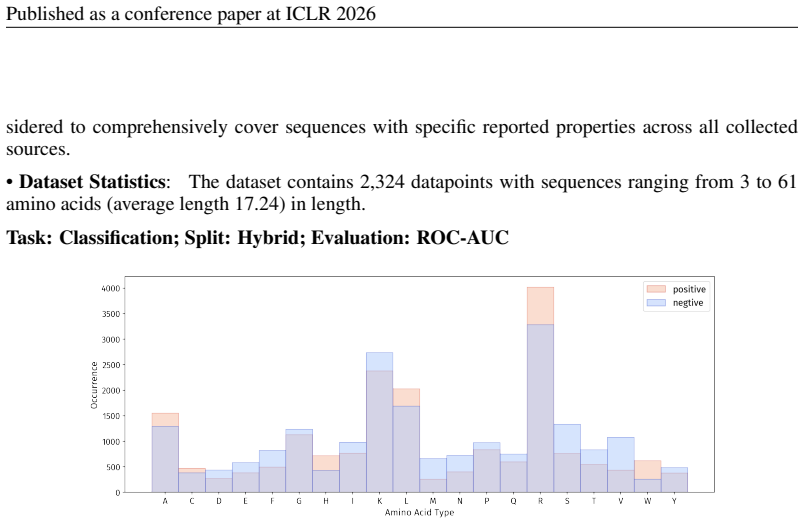
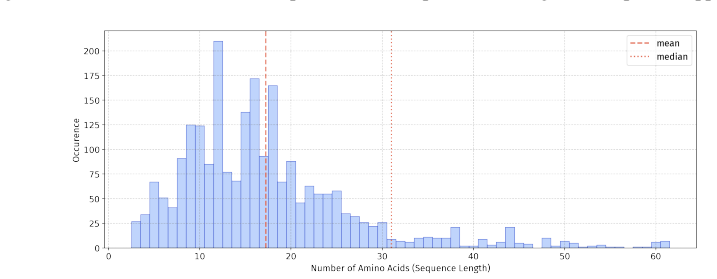
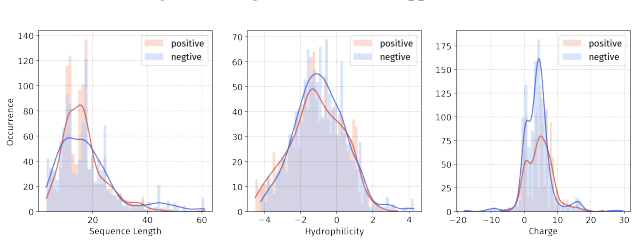
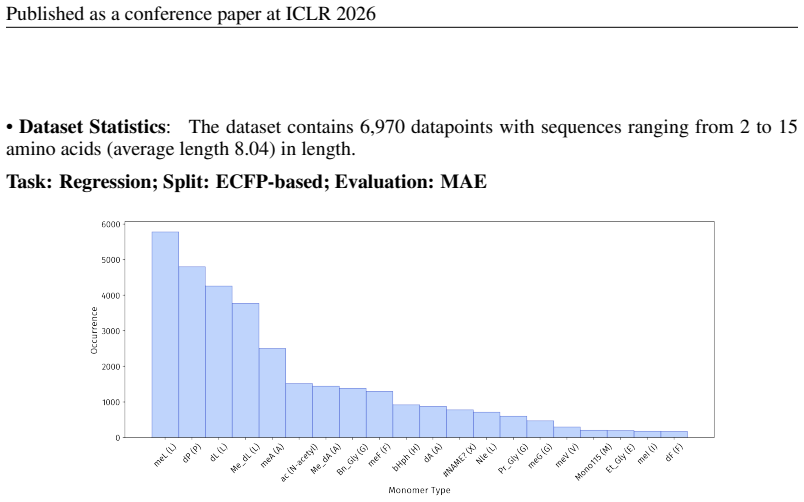
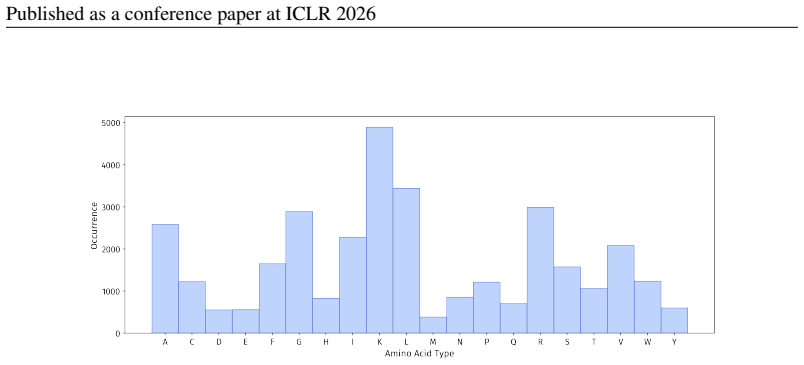
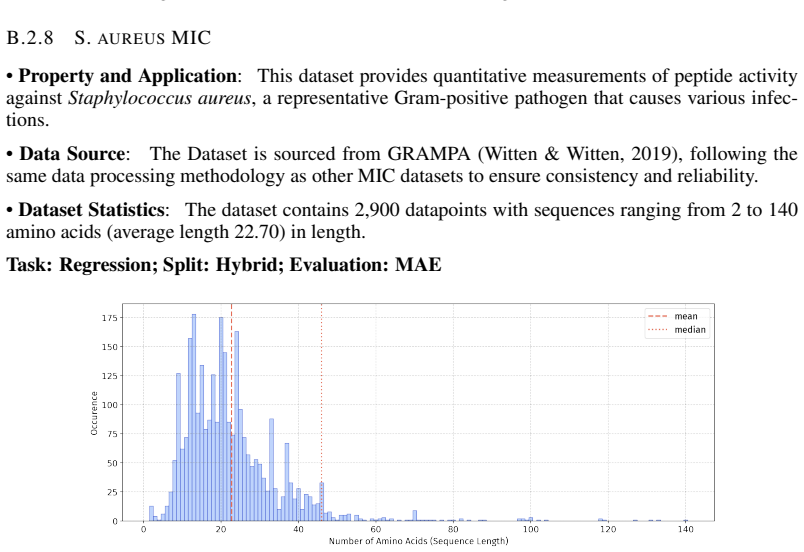
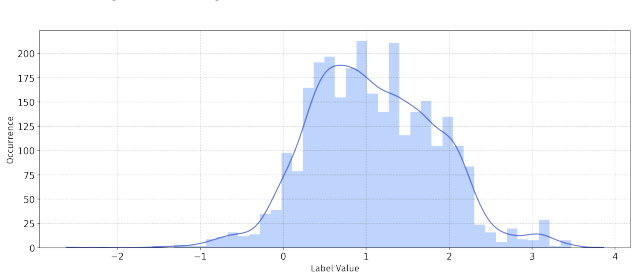
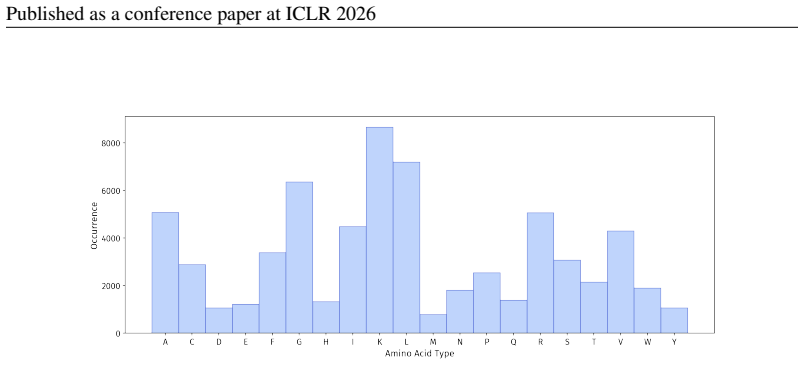
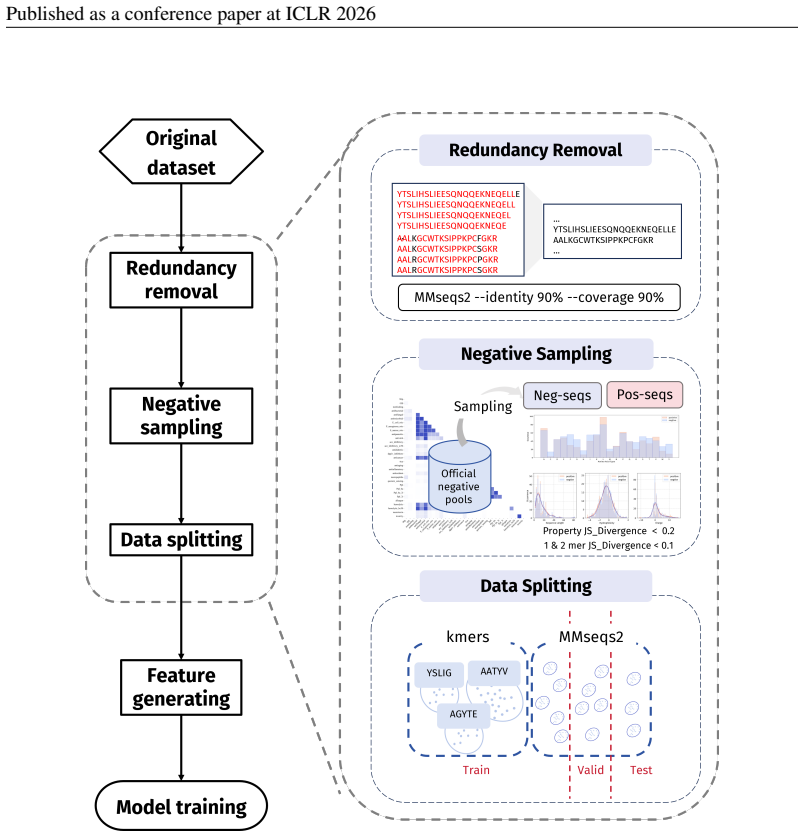
PepBenchmark comprises PepBenchData with 29 canonical and 6 non-canonical peptide datasets in 7 groups, PepBenchPipeline for standardized cleaning and splitting, and PepBenchLeaderboard with baselines across fingerprint, GNN, PLM, and SMILES model families, establishing a common foundation for peptide drug discovery.
What carries the argument
The PepBenchmark framework, which unifies data resources and protocols to enable consistent and comparable machine learning experiments on peptides.
If this is right
- Peptide ML methods can now be evaluated under identical conditions for direct comparison.
- Baselines from four methodological families provide reference points for new work.
- The coverage of both canonical and non-canonical peptides broadens applicability to diverse drug design problems.
- Standardized preprocessing reduces quality issues that arise in custom pipelines.
Where Pith is reading between the lines
- The availability of this resource may encourage more researchers to focus on peptide-specific modeling challenges rather than data preparation.
- Performance insights from the leaderboard could guide the selection of model architectures for particular peptide tasks.
- Over time, the benchmark might be expanded to incorporate additional real-world metrics such as experimental validation results.
Load-bearing premise
The 35 datasets and the preprocessing and splitting rules chosen for PepBenchPipeline are representative of real peptide drug-discovery challenges and free from systematic biases introduced by curation decisions.
What would settle it
An experiment showing that using different datasets or preprocessing steps outside PepBenchPipeline produces substantially altered model performance rankings would indicate that the benchmark does not fully capture the domain.
Figures

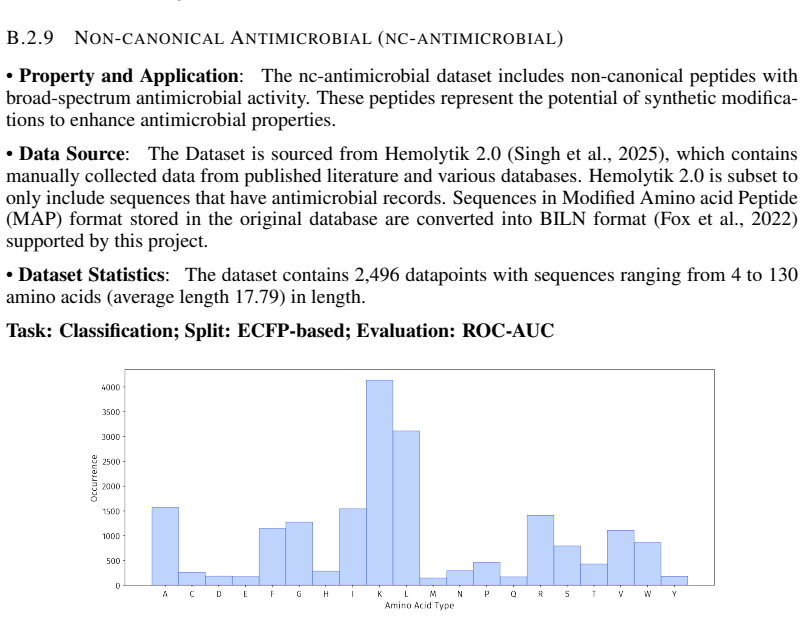

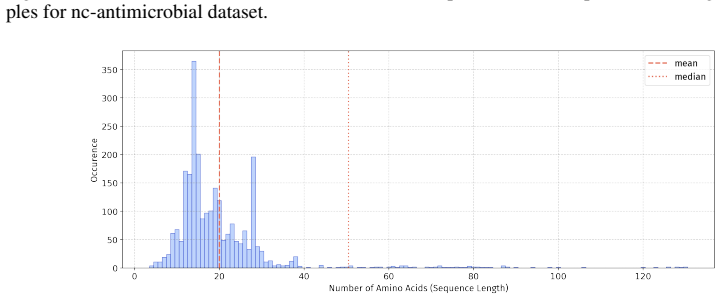
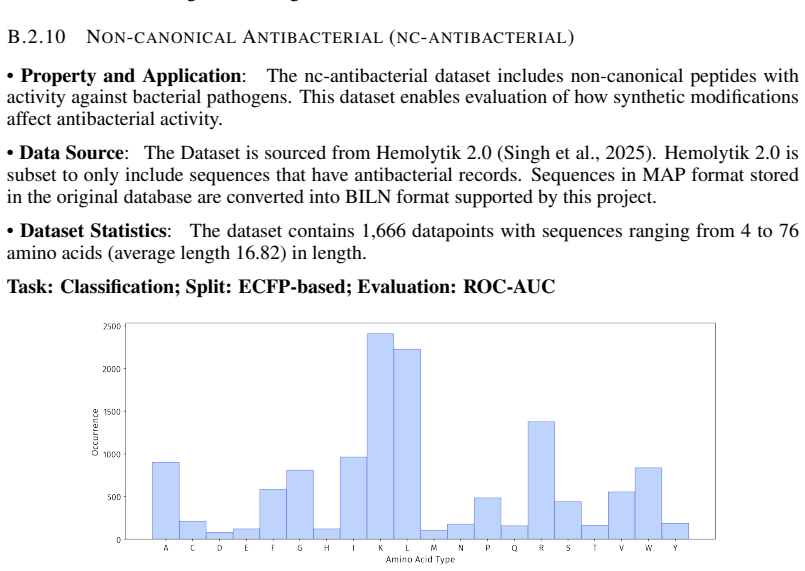

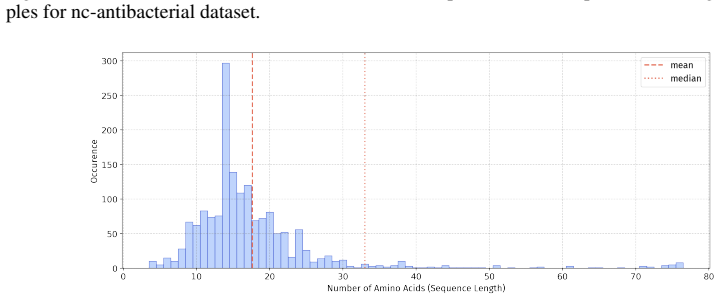





read the original abstract
Peptide therapeutics are widely regarded as the "third generation" of drugs, yet progress in peptide Machine Learning (ML) are hindered by the absence of standardized benchmarks. Here we present PepBenchmark, which unifies datasets, preprocessing, and evaluation protocols for peptide drug discovery. PepBenchmark comprises three components: (1) PepBenchData, a well-curated collection comprising 29 canonical-peptide and 6 non-canonical-peptide datasets across 7 groups, systematically covering key aspects of peptide drug development, representing, to the best of our knowledge, the most comprehensive AI-ready dataset resource to date; (2) PepBenchPipeline, a standardized preprocessing pipeline that ensures consistent dataset cleaning, construction, splitting, and feature transformation, mitigating quality issues common in ad hoc pipelines; and (3) PepBenchLeaderboard, a unified evaluation protocol and leaderboard with strong baselines across 4 major methodological families: Fingerprint-based, GNN-based, PLM-based, and SMILES-based models. Together, PepBenchmark provides the first standardized and comparable foundation for peptide drug discovery, facilitating methodological advances and translation into real-world applications. The data and code are publicly available at https://github.com/ZGCI-AI4S-Pep/PepBenchmark/.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces PepBenchmark as a unified resource for peptide ML in drug discovery, comprising PepBenchData (a curated set of 35 datasets: 29 canonical and 6 non-canonical across 7 groups), PepBenchPipeline (standardized preprocessing, cleaning, splitting, and feature transformation), and PepBenchLeaderboard (unified evaluation with baselines from fingerprint-based, GNN-based, PLM-based, and SMILES-based models). It claims this provides the first standardized and comparable foundation for the field, with public data and code release.
Significance. If the datasets prove representative of real-world peptide drug-discovery tasks and the pipeline avoids introducing curation biases, the benchmark could enable reproducible comparisons across methods and support translation to applications, analogous to MoleculeNet-style resources in other domains. The public release of datasets and code is a clear positive contribution.
major comments (2)
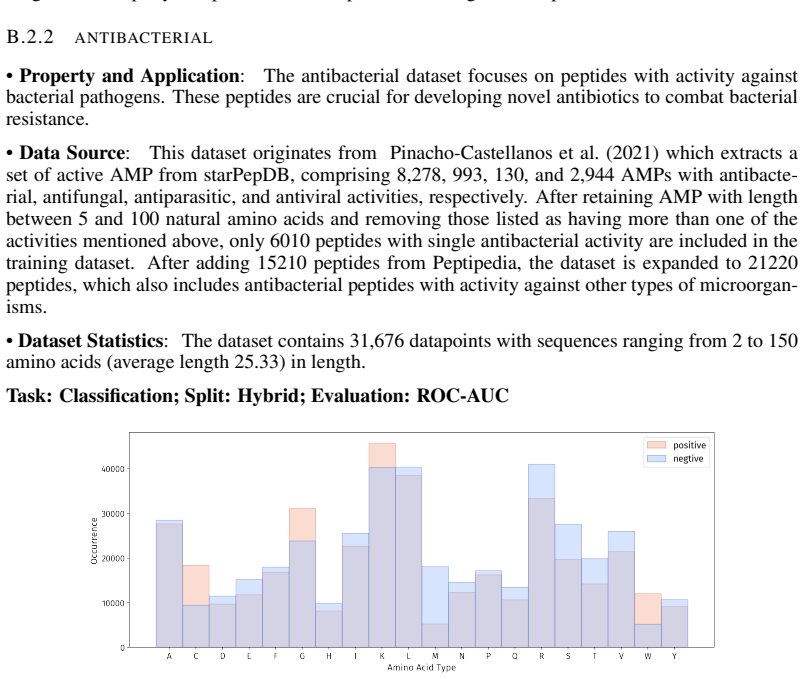
- [PepBenchData] PepBenchData section: The claim that the 35 datasets constitute 'the most comprehensive AI-ready dataset resource to date' and systematically cover key aspects of peptide drug development is asserted without quantitative validation. No statistical comparisons (e.g., sequence length distributions, modification frequencies, physicochemical property distributions, or task difficulty metrics) are provided against external references such as clinical peptide therapeutics databases or large peptide libraries, which is load-bearing for the 'standardized and comparable foundation' claim.
- [PepBenchPipeline] PepBenchPipeline description: The pipeline is presented as mitigating quality issues via systematic cleaning and splitting, yet no validation (e.g., ablation studies or checks confirming preservation of original task semantics) is reported. This leaves open the possibility that curation choices systematically bias the leaderboard results or limit generalizability to real discovery pipelines.
minor comments (2)
- The introduction could include explicit comparisons to prior benchmarks in related areas (e.g., protein or small-molecule ML) to sharpen the novelty statement.
- Clarify the precise selection criteria for the 7 groups and 35 datasets, including any exclusion rules, to improve reproducibility of the curation process.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and positive view of PepBenchmark's potential contribution. We address each major comment below and describe the revisions we will make.
read point-by-point responses
-
Referee: [PepBenchData] PepBenchData section: The claim that the 35 datasets constitute 'the most comprehensive AI-ready dataset resource to date' and systematically cover key aspects of peptide drug development is asserted without quantitative validation. No statistical comparisons (e.g., sequence length distributions, modification frequencies, physicochemical property distributions, or task difficulty metrics) are provided against external references such as clinical peptide therapeutics databases or large peptide libraries, which is load-bearing for the 'standardized and comparable foundation' claim.
Authors: We agree that quantitative validation would strengthen the claim. Our statement that PepBenchData is the most comprehensive AI-ready resource to date rests on a literature survey showing that prior peptide collections are smaller, task-specific, and lack unified preprocessing. The seven groups were deliberately chosen to span core peptide drug-development tasks (binding, toxicity, stability, etc.). In the revised manuscript we will add a dedicated subsection containing statistical comparisons of sequence-length distributions, modification frequencies, and physicochemical properties against accessible external references such as PepBank and curated clinical-peptide lists, thereby providing the requested quantitative support. revision: yes
-
Referee: [PepBenchPipeline] PepBenchPipeline description: The pipeline is presented as mitigating quality issues via systematic cleaning and splitting, yet no validation (e.g., ablation studies or checks confirming preservation of original task semantics) is reported. This leaves open the possibility that curation choices systematically bias the leaderboard results or limit generalizability to real discovery pipelines.
Authors: We appreciate the concern. The pipeline applies standard, widely adopted cleaning and splitting procedures to eliminate common ad-hoc artifacts and ensure reproducibility. We acknowledge that the original submission did not include explicit ablation or semantic-preservation checks. In the revised version we will add (i) ablation experiments quantifying the effect of each cleaning step on downstream model performance and (ii) distributional analyses demonstrating that the chosen splits preserve the original task semantics and label distributions. These additions will directly address the possibility of systematic bias. revision: yes
Circularity Check
No circularity; benchmark construction is explicit data aggregation and protocol definition with no derivations or self-referential reductions.
full rationale
The manuscript describes PepBenchmark as the assembly of 35 curated datasets (PepBenchData), a fixed preprocessing/splitting pipeline (PepBenchPipeline), and a unified leaderboard with baselines across model families (PepBenchLeaderboard). No equations, parameter fitting, predictions, or uniqueness theorems appear in the provided text. The claim of supplying the 'first standardized and comparable foundation' is an assertion of novelty and coverage rather than a derivation that reduces to its own inputs by construction. No self-citations are used to justify load-bearing steps, and the representativeness of the chosen datasets is presented as a curation choice rather than a fitted or self-defined result. The work is therefore self-contained as an engineering contribution.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The 29 canonical and 6 non-canonical datasets grouped into 7 categories collectively cover the key tasks in peptide drug development.
Reference graph
Works this paper leans on
-
[1]
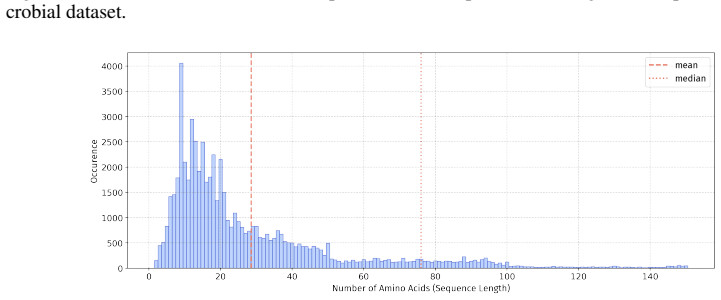
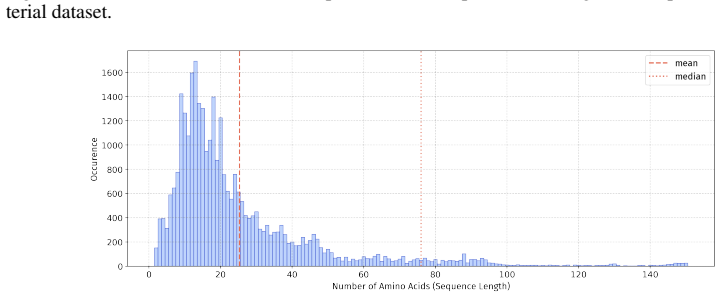
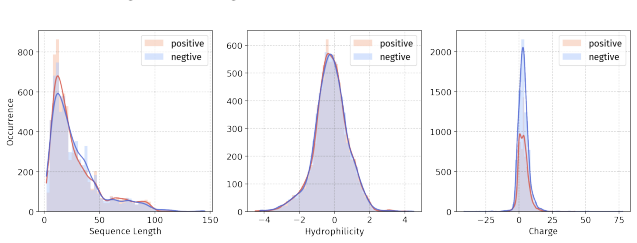
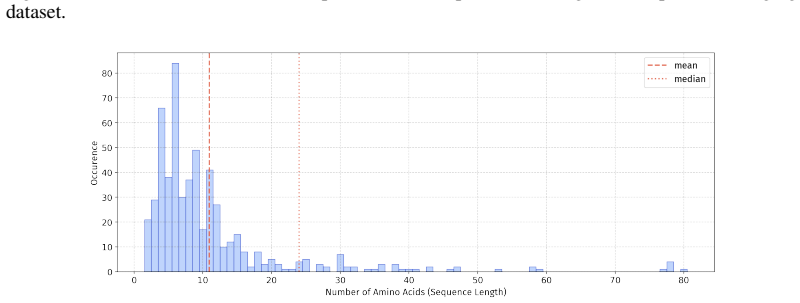
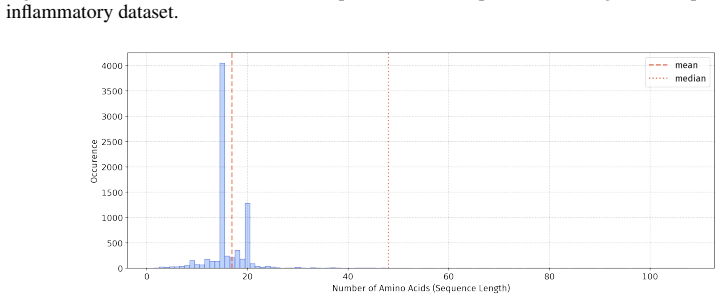
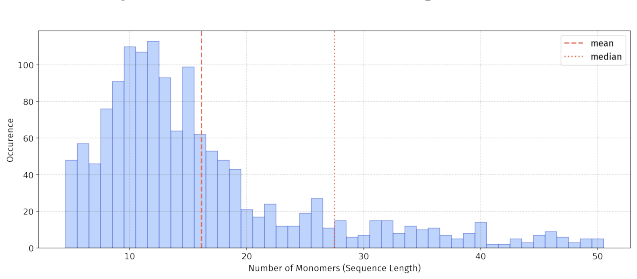
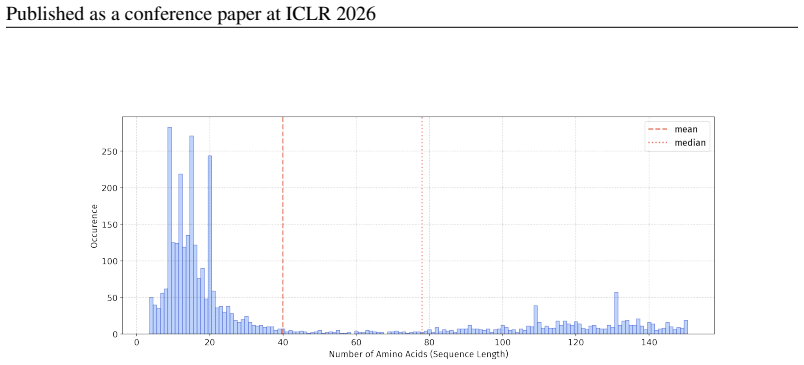
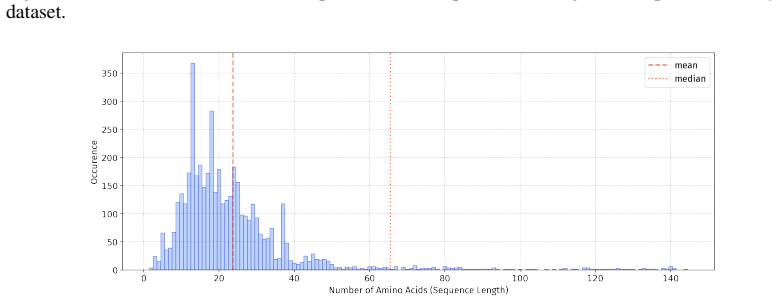
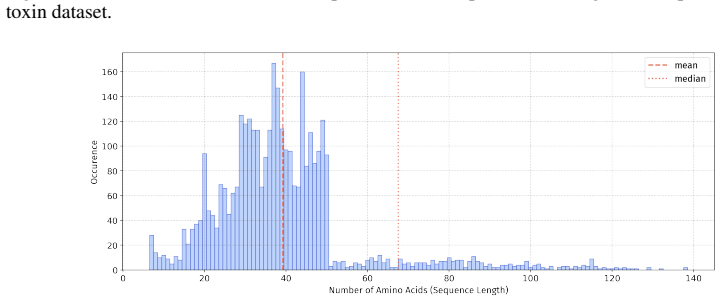
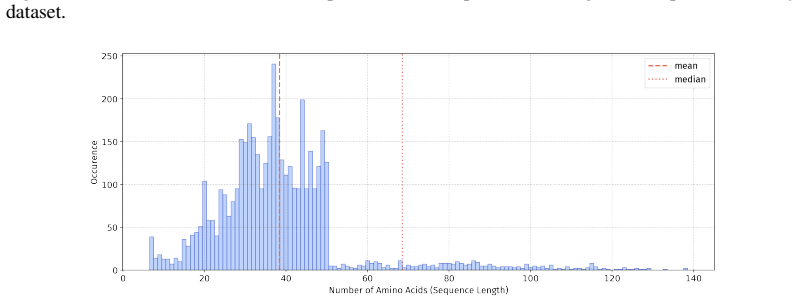
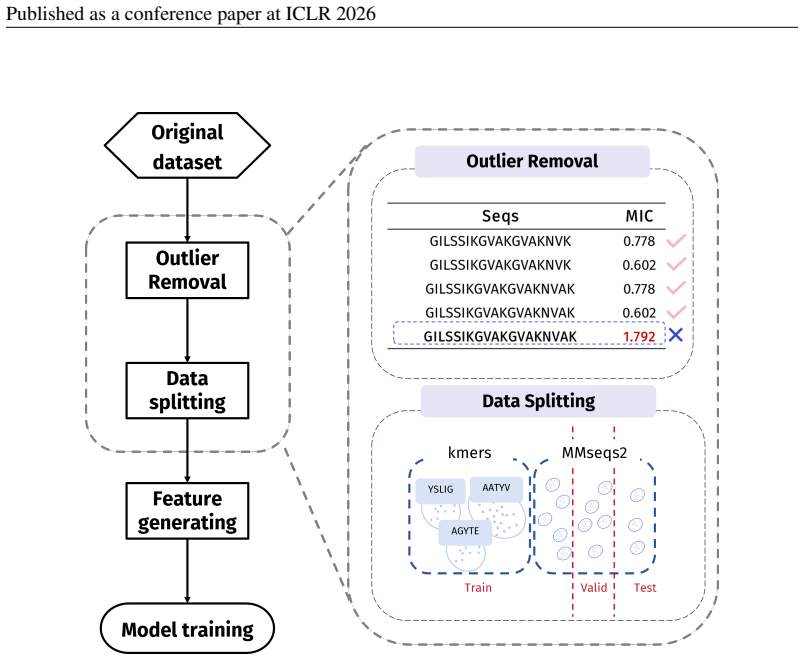
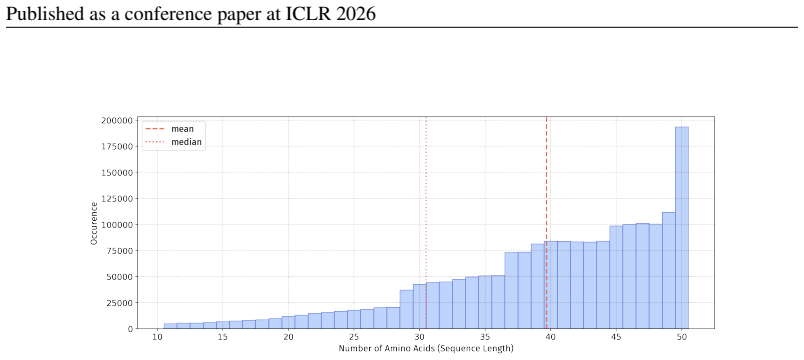
In 12 datasets, all sequences have lengths below 50
-
[2]
In 18 datasets, fewer than 10% of the sequences exceed a length of 50; 57 Published as a conference paper at ICLR 2026
work page 2026
-
[3]
For the benchmark, we believe it is necessary to define a unified length criterion
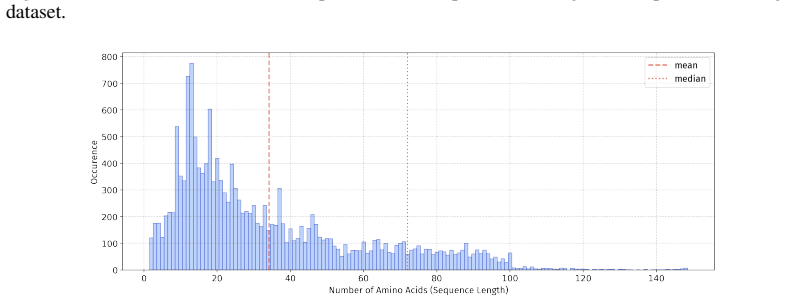
In 5 datasets, at least 10% of the sequences are longer than 50, with maximum lengths reaching up to 150 (including datasets on antifungal, antimicrobial, antiparasitic, anti- cancer, allergen, neurotoxin, and toxicity). For the benchmark, we believe it is necessary to define a unified length criterion. From the perspective of drug development experts, di...
work page 2019
-
[4]
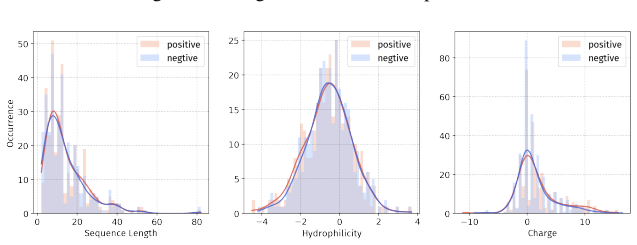
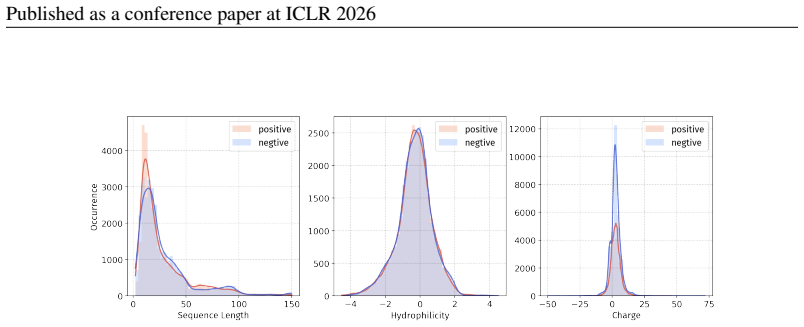
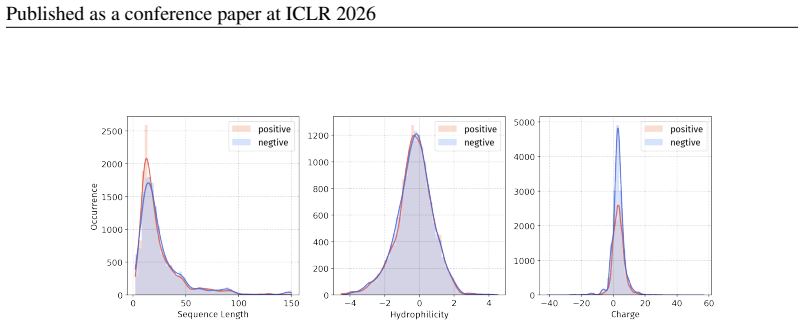
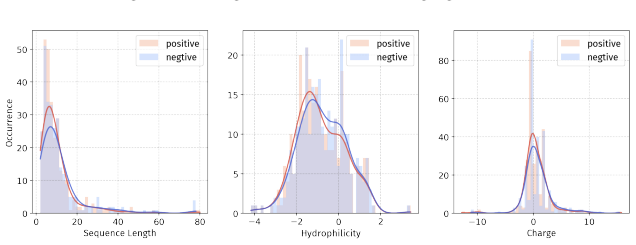
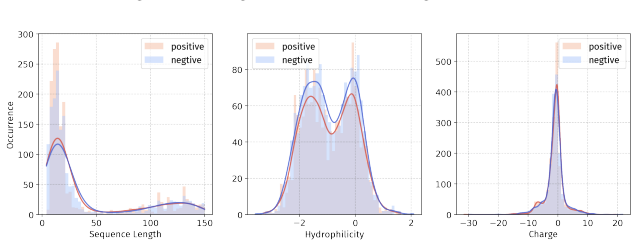
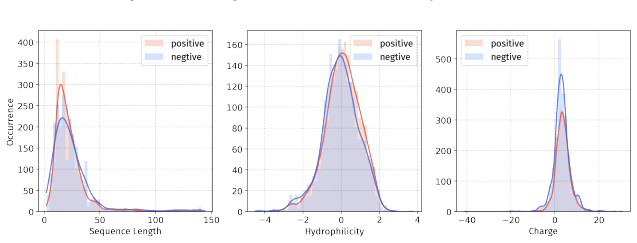
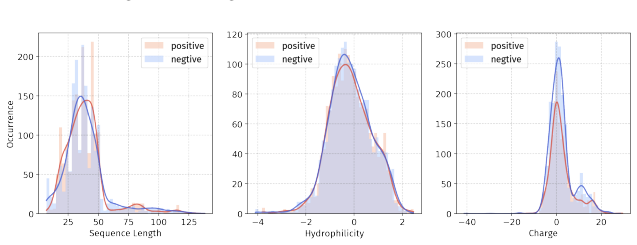
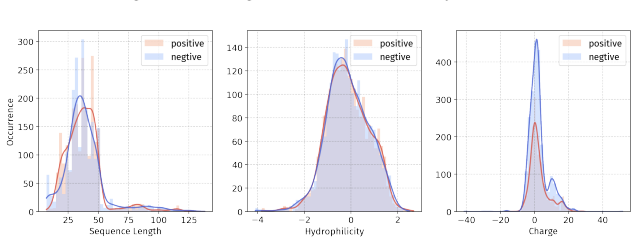
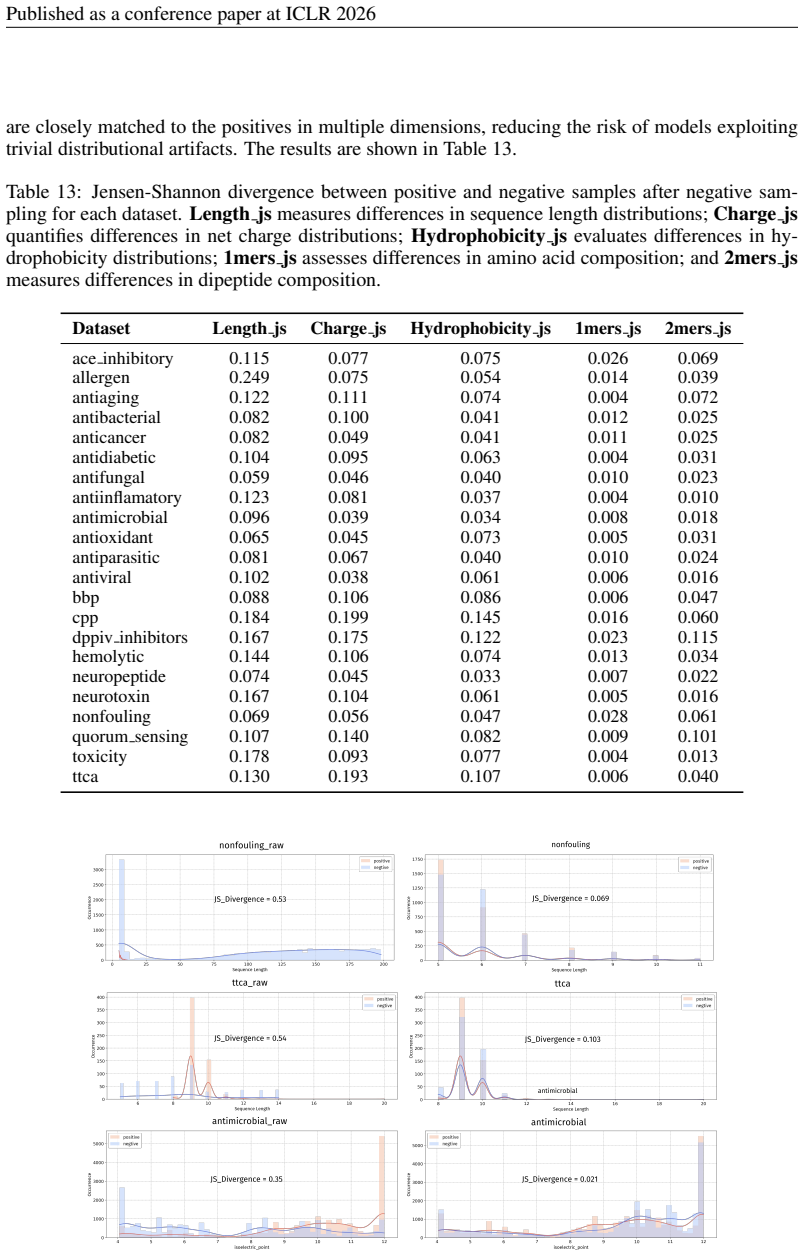
Principle 1: The differences between positive and negative samples should reflect gener- alizable biological properties rather than dataset-specific artifacts (e.g., clear distributional differences, inherent contrasts between active and inactive peptides)
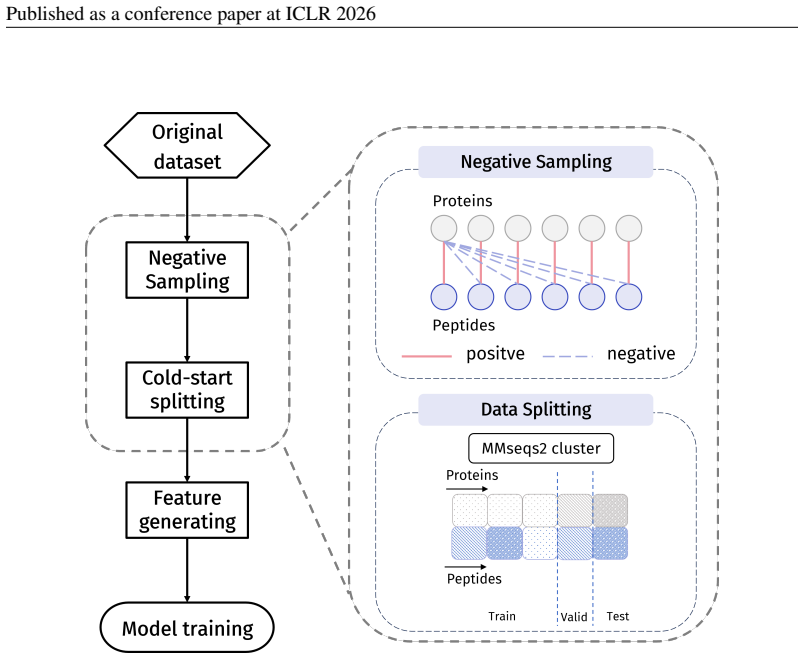
-
[5]
Principle 2·: False negatives should be avoided as much as possible. Based on these principles, we introduceBiologically Informed and Distribution-Controlled Neg- ative Sampling (BDNegSamp). The procedure involves three key steps: Step 1: Construction of the Biologically Informed Negative Sample PoolThe initial negative pool is defined as all collected bi...
-
[6]
Run the enrichment analysis to identify significantly enriched k-mers (henceforthmotifs)
-
[7]
Group sequences into clusters such that any two sequences sharing at least one enriched motif are placed in the same cluster
-
[8]
Assign clusters (not sequences) to train/validation/test according to the target ratio (8:1:1 in our experiments). By construction, this protocol prevents any enriched motif from appearing across partitions, thereby forcing models to generalize beyond dataset-specific shortcuts and yielding a more faithful estimate of cross-motif generalization difficulty...
-
[9]
Apply the kmer–aware procedure in subsubsection D.4.1 to identify motif clusters and allocate them to splits
-
[10]
For MMseqs2-based homology splitting, parameter settings also need to be standardized
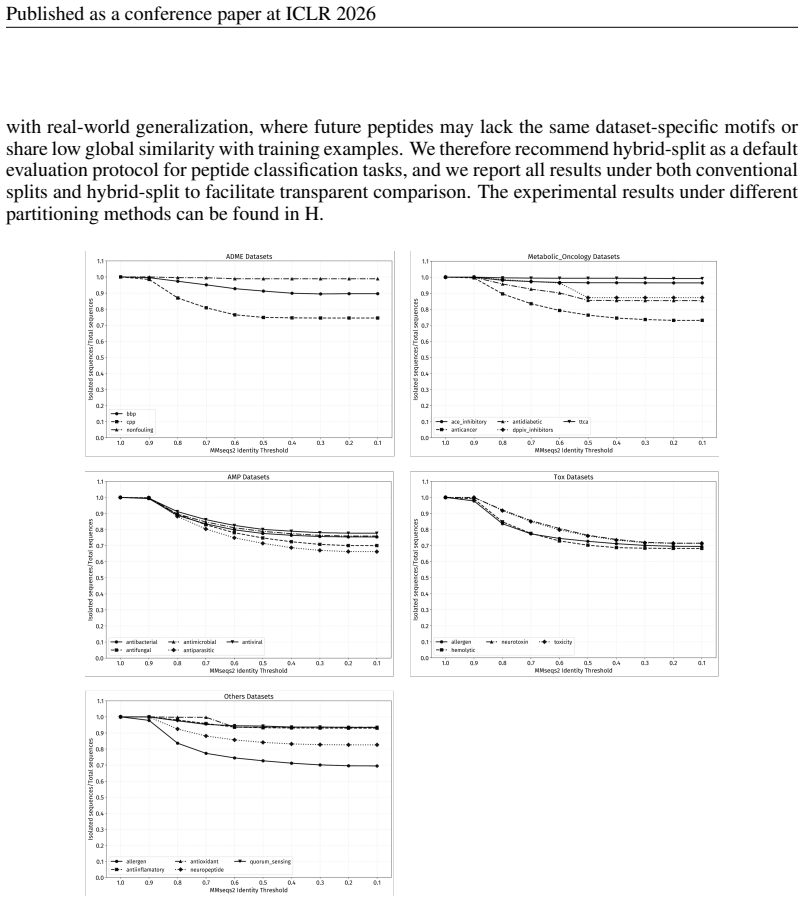
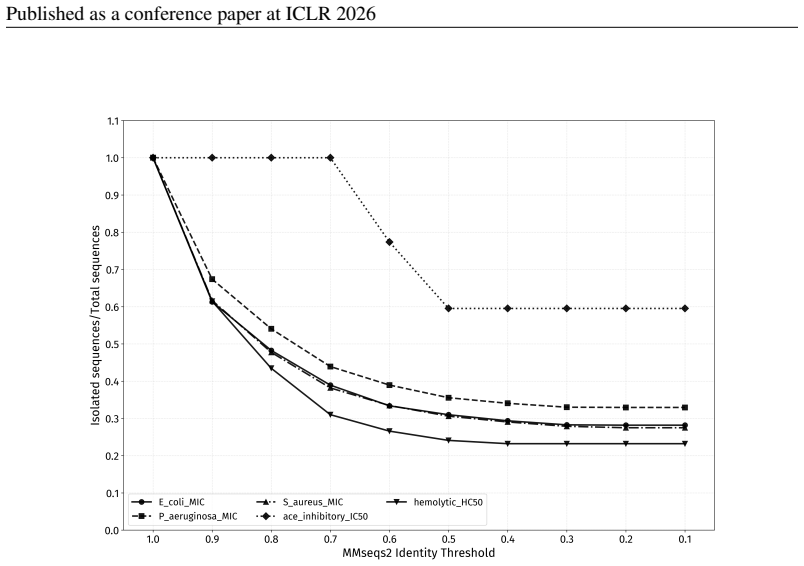
For sequences that do not contain any enriched motif, applyMMseqs2to form homology clusters and allocate these clusters to the existing splits, maintaining the desired propor- tions. For MMseqs2-based homology splitting, parameter settings also need to be standardized. By ex- amining the changes in the number of isolated sequences under different identity...
work page 2026
-
[11]
Results on 22 canonical peptide classification datasets under four splitting strategies: hybrid-split(Table 21),MMseqs2-split(Table 23),k-mer–split(Table 22), andrandom- split(Table 24)
-
[12]
Results on 4 non-canonical peptide regression datasets under four splitting strategies: hybrid-split(Table 25),MMseqs2-split(Table 27),k-mer–split(Table 26), andrandom- split(Table 28)
-
[13]
Results on 5 non-canonical peptide datasets under five splitting strategies:ECFP-split(Ta- ble 29),hybrid-split(Table 30),MMseqs2-split(Table 32),k-mer–split(Table 31), and random-split(Table 33)
-
[14]
Results on 3 PepPI datasets without freezing the protein encoder (Table 34)
-
[15]
Results on thePepBenchData-150benchmark (Table 36). For most datasets in PepBenchData-150, the proportion of sequences longer than 50 residues is below 10% (see Table 7 and Table 8). Therefore, we consider these datasets to be largely compara- ble to thePepBenchData-50version. Only five datasets contain more than 50% sequences longer than 50 residues, and...
-
[16]
The conclusions in the main text hold across all partitioning strategies
-
[17]
The performance of kmer-split is significantly lower than that of random-split, indicating that the issue of kmer leakage is severe. The hybrid partition combines the advantages of MMseqs2-split and kmer-split, making it a more challenging partitioning strategy
-
[18]
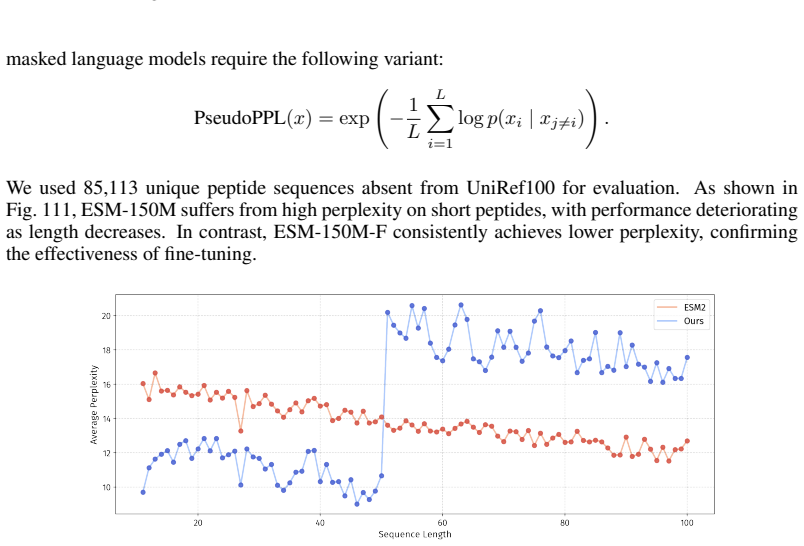
In the PepBenchData-150 version, the fine-tuned ESM2-150-F performs worse than ESM2- 150M. This result is expected, as ESM2-150-F is fine-tuned only on peptide data and thus forgets the protein pre-training knowledge. As shown in Figure 111, ESM2-150M-F ex- hibits higher perplexity than ESM2-150M on sequences longer than 50
-
[19]
bbp", official_feature_names=[
For the PepPI task, it remains uncertain whether freezing the protein encoder is necessary. 74 Published as a conference paper at ICLR 2026 Table 21: Performance of models on canonical peptide classification (ROC-AUC↑, %) with hybrid- split. Dataset sizes are shown separately; results are mean±std. Best and second-best scores per row are inboldand gray sh...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.