Recognition: unknown
Polyformer: a generative framework for thermodynamic modeling of polymeric molecules
Pith reviewed 2026-05-10 12:32 UTC · model grok-4.3
The pith
Polyformer generates biomolecular conformations that match temperature-dependent thermodynamic ensembles from sequence input alone.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Given a sequence and temperature, the Polyformer produces conformations faithful to the molecule's thermodynamic ensemble. It thereby addresses molecular folding, the statistics of the conformational ensemble, and the explicit dependence of that ensemble on temperature within a single generative model trained on molecular dynamics data.
What carries the argument
A conditional generative model that maps sequence and thermodynamic variable directly to samples from the equilibrium conformational distribution.
If this is right
- The model supplies both individual structures and ensemble statistics in one forward pass.
- Temperature enters as an explicit input, so conformational populations can be queried at any chosen temperature without new simulations.
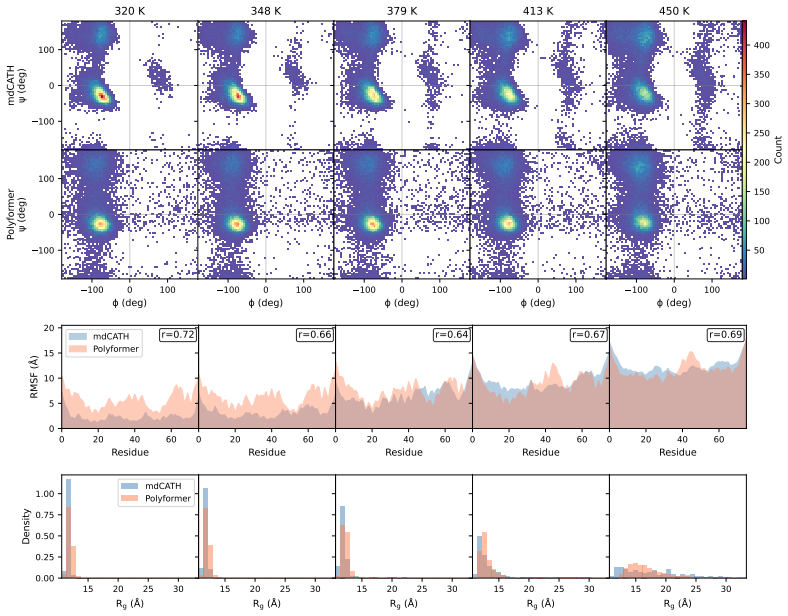
- Direct comparison to molecular dynamics trajectories shows agreement for protein domains of 50-111 residues.
Where Pith is reading between the lines
- If the model generalizes, it could let researchers explore how sequence changes alter ensemble properties rather than just the lowest-energy fold.
- The same conditioning approach might extend to other polymeric systems such as nucleic acids or synthetic chains once suitable training trajectories exist.
- One concrete test would be to measure agreement between model outputs and experimental ensemble observables such as NMR order parameters at multiple temperatures.
Load-bearing premise
A model trained on molecular dynamics trajectories for a limited set of protein domains will produce faithful ensembles for arbitrary sequences and temperatures outside the training data.
What would settle it
For a sequence and temperature withheld from training, the statistical properties of conformations generated by the model (such as radius of gyration distribution or secondary-structure content) differ markedly from those obtained in independent, long molecular dynamics runs.
Figures













read the original abstract
The classic paradigm of structural biology is that the sequence of a biomolecule (protein, nucleic acid, lipid, etc) determines its conformation (shape) which determines its biological function. Protein folding programs like AlphaFold address this paradigm by predicting the single best conformation given a sequence that defines the molecule. However, biomolecules are not static structures, and their conformational ensemble determines their function. We present the Polyformer -- a generative framework for thermodynamic modeling of polymeric molecules. Given the sequence and temperature (or another thermodynamic variable), the Polyformer generates conformations faithful to the molecule's thermodynamic conformational ensemble. It is the first generative model that solves three problems simultaneously: how does a molecule fold, what is its conformational ensemble, and how does the conformational ensemble change as we change physical temperature. As a concrete test case, we apply Polyformer to protein domains with 50-111 residues and report good agreement of model predictions to Molecular Dynamics (MD) trajectories.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces Polyformer, a generative framework that takes a polymeric molecule's sequence and a thermodynamic variable (e.g., temperature) as input and generates conformations sampled from the molecule's thermodynamic ensemble. It claims to be the first model to simultaneously solve molecular folding, conformational ensemble generation, and temperature dependence of the ensemble, with a concrete demonstration on protein domains of 50-111 residues reporting good agreement with molecular dynamics trajectories.
Significance. If the model can be shown to produce faithful Boltzmann ensembles for sequences and temperatures outside the training distribution without circular reproduction of MD data, it would constitute a meaningful advance in computational structural biology by extending beyond static predictors such as AlphaFold to a unified, temperature-aware generative treatment of conformational thermodynamics.
major comments (2)
- [Abstract] Abstract: the central claim of 'good agreement' with MD trajectories for 50-111 residue domains is unsupported by any quantitative metrics, error bars, training details, or validation protocol, which prevents assessment of whether the outputs reflect thermodynamic ensembles or learned distributions.
- [Abstract] Abstract: no information is supplied on the temperature range used for training, whether test temperatures were held out, or whether sequences were disjoint from the training set; without such controls the claimed temperature-dependence component reduces to interpolation rather than a general thermodynamic model.
Simulated Author's Rebuttal
We thank the referee for their constructive review of our manuscript. We address each major comment below and have revised the manuscript to strengthen the abstract with additional details.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim of 'good agreement' with MD trajectories for 50-111 residue domains is unsupported by any quantitative metrics, error bars, training details, or validation protocol, which prevents assessment of whether the outputs reflect thermodynamic ensembles or learned distributions.
Authors: We agree that the abstract would benefit from explicit quantitative support. We will revise the abstract to include key quantitative metrics of agreement with MD trajectories (such as average RMSD and ensemble overlap statistics with associated error bars) along with a concise summary of the validation protocol. This change will allow readers to directly evaluate the thermodynamic fidelity of the generated ensembles. revision: yes
-
Referee: [Abstract] Abstract: no information is supplied on the temperature range used for training, whether test temperatures were held out, or whether sequences were disjoint from the training set; without such controls the claimed temperature-dependence component reduces to interpolation rather than a general thermodynamic model.
Authors: We agree that these controls are essential to substantiate the temperature-dependence claim. We will revise the abstract to explicitly state the temperature range used during training, confirm that test temperatures were held out, and note that sequences were disjoint from the training set. This will clarify the distinction between interpolation and generalization in the model's thermodynamic predictions. revision: yes
Circularity Check
No circularity in claimed derivation
full rationale
The paper presents Polyformer as a data-driven generative model trained on MD trajectories to sample conformational ensembles conditioned on sequence and temperature. No mathematical derivation chain, first-principles equations, or self-referential definitions appear in the abstract or described claims. The reported agreement with MD is an empirical evaluation of a learned distribution rather than a prediction forced by construction from the same inputs. No self-citation load-bearing steps, uniqueness theorems, or ansatz smuggling are identified. The work is self-contained as a standard ML framework whose validity rests on external MD benchmarks, not internal reduction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Protein conformational ensembles in function: roles and mechanisms.RSC Chem
Ruth Nussinov, Yonglan Liu, Wengang Zhang, and Hyunbum Jang. Protein conformational ensembles in function: roles and mechanisms.RSC Chem. Biol., 4:850–864, 2023
2023
-
[2]
P. G. de Gennes. Conformations of polymers attached to an interface.Macromolecules, 13(5):1069–1075, 09 1980. 9
1980
-
[3]
Wright and H
Peter E. Wright and H. Jane Dyson. Intrinsically disordered proteins in cellular signalling and regulation.Nature Reviews Molecular Cell Biology, 16(1):18–29, 2015
2015
-
[4]
Large-scale conformational changes and protein function: Breaking the in silico barrier.Frontiers in Molecular Biosciences, V olume 6 - 2019, 2019
Laura Orellana. Large-scale conformational changes and protein function: Breaking the in silico barrier.Frontiers in Molecular Biosciences, V olume 6 - 2019, 2019
2019
-
[5]
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, Alex Bridgland, Clemens Meyer, Simon A. A. Kohl, Andrew J. Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reim...
2021
-
[6]
Kinch, R
Minkyung Baek, Frank DiMaio, Ivan Anishchenko, Justas Dauparas, Sergey Ovchinnikov, Gyu Rie Lee, Jue Wang, Qian Cong, Lisa N. Kinch, R. Dustin Schaeffer, Claudia Millán, Hahnbeom Park, Carson Adams, Caleb R. Glassman, Andy DeGiovanni, Jose H. Pereira, Andria V . Rodrigues, Alberdina A. van Dijk, Ana C. Ebrecht, Diederik J. Opperman, Theo Sagmeister, Chris...
2021
-
[7]
Alphafold meets flow matching for generating protein ensembles, 2024
Bowen Jing, Bonnie Berger, and Tommi Jaakkola. Alphafold meets flow matching for generating protein ensembles, 2024
2024
-
[8]
Simplefold: Folding proteins is simpler than you think
Yuyang Wang, Jiarui Lu, Navdeep Jaitly, Josh Susskind, and Miguel Angel Bautista. Simplefold: Folding proteins is simpler than you think. InICLR, 2025
2025
-
[9]
Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model.Science, 379(6637):1123–1130, 2023
2023
-
[10]
mdcath: A large-scale md dataset for data-driven computational biophysics.Scientific Data, 11(1):1299, 2024
Antonio Mirarchi, Toni Giorgino, and Gianni De Fabritiis. mdcath: A large-scale md dataset for data-driven computational biophysics.Scientific Data, 11(1):1299, 2024
2024
-
[11]
Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning.Science, 365(6457):eaaw1147, 2019
Frank Noé, Simon Olsson, Jonas Köhler, and Hao Wu. Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning.Science, 365(6457):eaaw1147, 2019
2019
-
[12]
From data to noise to data for mixing physics across temperatures with generative artificial intelligence.Proceedings of the National Academy of Sciences, 119(32):e2203656119, 2022
Yihang Wang, Lukas Herron, and Pratyush Tiwary. From data to noise to data for mixing physics across temperatures with generative artificial intelligence.Proceedings of the National Academy of Sciences, 119(32):e2203656119, 2022
2022
-
[13]
I: The new millennium edition: mainly mechanics, radiation, and heat, volume 1
Richard P Feynman, Robert B Leighton, and Matthew Sands.The Feynman lectures on physics, Vol. I: The new millennium edition: mainly mechanics, radiation, and heat, volume 1. Basic books, 2011
2011
-
[14]
Ballard, Joshua Bambrick, Sebastian W
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J. Ballard, Joshua Bambrick, Sebastian W. Bodenstein, David A. Evans, Chia-Chun Hung, Michael O’Neill, David Reiman, Kathryn Tunyasuvunakool, Zachary Wu, Akvil˙e Žemgulyt ˙e, Eirini Arvaniti, Charles Beattie, Ottavia Bertolli, A...
2024
-
[15]
Boltz-1 democratizing biomolecular interaction modeling.bioRxiv, 2025
Jeremy Wohlwend, Gabriele Corso, Saro Passaro, Noah Getz, Mateo Reveiz, Ken Leidal, Wojtek Swiderski, Liam Atkinson, Tally Portnoi, Itamar Chinn, Jacob Silterra, Tommi Jaakkola, and Regina Barzilay. Boltz-1 democratizing biomolecular interaction modeling.bioRxiv, 2025
2025
-
[16]
Deep generative modeling of temperature-dependent structural ensembles of proteins.Communications Chemistry, 8(1):354, 2025
Giacomo Janson, Alexander Jussupow, and Michael Feig. Deep generative modeling of temperature-dependent structural ensembles of proteins.Communications Chemistry, 8(1):354, 2025
2025
-
[17]
Schneekloth, and Pratyush Tiwary
Lukas Herron, Yunrui Qiu, Anjali Verma, Venkata Sai Sreyas Adury, Richard John, Suemin Lee, Shams Mehdi, Disha Sanwal, John S. Schneekloth, and Pratyush Tiwary. Ab initio prediction of rna structure ensembles with rnanneal.bioRxiv, 2026
2026
-
[18]
Degiacomi, and Chris G
Adam Leach, Sebastian M Schmon, Matteo T. Degiacomi, and Chris G. Willcocks. Denoising diffusion probabilistic models on SO(3) for rotational alignment. InICLR 2022 Workshop on Geometrical and Topological Representation Learning, 2022
2022
-
[19]
Scalable diffusion models with transformers, 2023
William Peebles and Saining Xie. Scalable diffusion models with transformers, 2023
2023
-
[20]
Decoupled weight decay regularization, 2019
Ilya Loshchilov and Frank Hutter. Decoupled weight decay regularization, 2019
2019
-
[21]
Learnable fourier features for multi-dimensional spatial positional encoding.Advances in Neural Information Processing Systems, 34:15816–15829, 2021
Yang Li, Si Si, Gang Li, Cho-Jui Hsieh, and Samy Bengio. Learnable fourier features for multi-dimensional spatial positional encoding.Advances in Neural Information Processing Systems, 34:15816–15829, 2021
2021
-
[22]
Replica-exchange molecular dynamics method for protein folding.Chemical physics letters, 314(1-2):141–151, 1999
Yuji Sugita and Yuko Okamoto. Replica-exchange molecular dynamics method for protein folding.Chemical physics letters, 314(1-2):141–151, 1999
1999
-
[23]
Nonphysical sampling distributions in monte carlo free- energy estimation: Umbrella sampling.Journal of computational physics, 23(2):187–199, 1977
Glenn M Torrie and John P Valleau. Nonphysical sampling distributions in monte carlo free- energy estimation: Umbrella sampling.Journal of computational physics, 23(2):187–199, 1977
1977
-
[24]
Unsupervised generative modeling using matrix product states.Physical Review X, 8(3):031012, 2018
Zhao-Yu Han, Jun Wang, Heng Fan, Lei Wang, and Pan Zhang. Unsupervised generative modeling using matrix product states.Physical Review X, 8(3):031012, 2018
2018
-
[25]
Charmm36m: an improved force field for folded and intrinsically disordered proteins.Nature methods, 14(1):71–73, 2017
Jing Huang, Sarah Rauscher, Grzegorz Nawrocki, Ting Ran, Michael Feig, Bert L De Groot, Helmut Grubmüller, and Alexander D MacKerell Jr. Charmm36m: an improved force field for folded and intrinsically disordered proteins.Nature methods, 14(1):71–73, 2017
2017
-
[26]
Efficient diffusion training via min-snr weighting strategy
Tiankai Hang, Shuyang Gu, Chen Li, Jianmin Bao, Dong Chen, Han Hu, Xin Geng, and Baining Guo. Efficient diffusion training via min-snr weighting strategy. InProceedings of the IEEE/CVF international conference on computer vision, pages 7441–7451, 2023
2023
-
[27]
Denoising Diffusion Implicit Models
Jiaming Song, Chenlin Meng, and Stefano Ermon. Denoising diffusion implicit models.arXiv preprint arXiv:2010.02502, 2020. 11 Appendix A Forward diffusion Translation noise (VP-SDE, linear β).Translations are first scaled by a coordinate scaling factor s= 0.1 to bring Ångström coordinates into unit range. The instantaneous noise rate is β(t) = βmin +t(β ma...
work page internal anchor Pith review Pith/arXiv arXiv 2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.