Predicting Solvation Free Energies of Molecules and Ions via First-Principles and Machine-Learning Molecular Dynamics
Pith reviewed 2026-05-10 06:40 UTC · model grok-4.3
The pith
The bubble method computes solvation free energies for molecules and ions from first-principles molecular dynamics without end-state singularities or empirical data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Here, we introduce the bubble method to calculate the SFEs of molecules and ions from first principles. Our approach avoids the end-state problem in both ab initio and machine learning molecular dynamics simulations and is applicable to molecules and ions of arbitrary shape. When calculating the SFEs of ions using periodic density functional theory, we incorporate corrections for the neutralizing background charge, spurious interactions between periodic images, and the vacuum-water interface potential. To validate our method, we successfully computed the SFEs of methane, methanol, water, and sodium ions using classical, ab initio, and machine learning molecular dynamics simulations. Our work
What carries the argument
The bubble method, which forms a spherical cavity in the solvent and gradually populates it with the solute to compute the reversible work of insertion while avoiding close-contact singularities.
Load-bearing premise
The bubble method correctly sidesteps the end-state singularity for arbitrary shapes and the periodic-boundary corrections for ions yield accurate first-principles SFEs without hidden empirical adjustments or insufficient sampling.
What would settle it
A large discrepancy between the sodium ion solvation free energy computed via the bubble method in ab initio MD and the established experimental value after all periodic corrections have been applied.
Figures



read the original abstract
The solvation free energy (SFE) of molecules and ions is a fundamental property governing their solvation behavior and solubility. Molecular simulations offer a route to compute SFEs using alchemical free energy methods, such as thermodynamic integration or free energy perturbation. However, these methods suffer from the infamous end-point singularity, which leads to numerical instability when atoms approach closely, a challenge that becomes particularly acute in ab initio and machine learning molecular dynamics simulations. Here, we introduce the bubble method to calculate the SFEs of molecules and ions from first principles. Our approach avoids the end-state problem in both ab initio and machine learning molecular dynamics simulations and is applicable to molecules and ions of arbitrary shape. When calculating the SFEs of ions using periodic density functional theory, we incorporate corrections for the neutralizing background charge, spurious interactions between periodic images, and the vacuum-water interface potential. To validate our method, we successfully computed the SFEs of methane, methanol, water, and sodium ions using classical, ab initio, and machine learning molecular dynamics simulations. Importantly, our method requires no experimental inputs or empirical data. This makes it particularly well-suited for studying systems under extreme conditions, such as high pressure-temperature environments or under nanoconfinement, situations where experimental investigations are challenging and classical force fields, typically parameterized under ambient conditions, may be unreliable.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the 'bubble method' to compute solvation free energies (SFEs) of molecules and ions from first principles via molecular dynamics. The approach replaces standard alchemical transformations with a bubble construction that avoids the end-state singularity and applies to solutes of arbitrary shape. For ions, three periodic-boundary corrections are added: neutralizing background charge, spurious periodic-image interactions, and the vacuum-water interface potential. Validation consists of SFE calculations for methane, methanol, water, and Na+ performed with classical, ab initio, and machine-learning MD trajectories, with the explicit claim that the procedure requires no experimental inputs or empirical parameters.
Significance. If the bubble construction and the three ion corrections can be shown to be robust and free of hidden empirical choices, the method would enable parameter-free SFE predictions under extreme conditions (high P-T, nanoconfinement) where classical force fields are unreliable and experiments are difficult, thereby strengthening first-principles routes to solvation thermodynamics.
major comments (3)
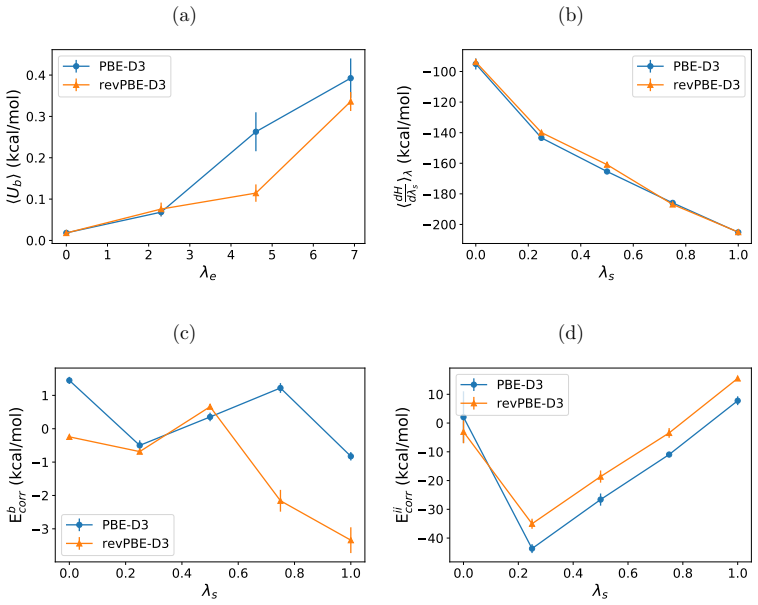
- [Bubble Method section] The central claim that the bubble method eliminates the end-state singularity for non-spherical solutes (methanol, water) while maintaining sufficient phase-space overlap rests on the alchemical integration along the bubble coordinate; however, the manuscript supplies no overlap metrics, lambda schedules, or variance estimates for the AIMD and MLMD runs, leaving open the possibility that the reported success is limited to well-behaved spherical cases.
- [Ion Corrections subsection] For the ion corrections, the vacuum-water interface potential term is known to be sensitive to slab thickness, water-model details, and reference-potential choice; the text does not report convergence tests with respect to these parameters or demonstrate that the chosen value is independent of classical data, which directly undermines the assertion that the entire procedure is purely first-principles.
- [Results section] The validation is restricted to four small species; because the interface-potential correction and bubble construction must remain accurate when solute charge or shape changes, the limited test set does not yet establish the general applicability required by the abstract.
minor comments (2)
- The abstract asserts that SFEs were 'successfully computed' yet contains no numerical values, error bars, or comparison to literature; adding at least one representative result would strengthen the summary.
- Notation for the bubble radius and the precise definition of the alchemical path should be clarified with an equation to avoid ambiguity when readers attempt to reproduce the integration.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review of our manuscript. We address each major comment point by point below, providing the strongest honest defense of the work while acknowledging where revisions are warranted. We have prepared a revised manuscript incorporating changes where they strengthen the presentation without misrepresenting the original results.
read point-by-point responses
-
Referee: [Bubble Method section] The central claim that the bubble method eliminates the end-state singularity for non-spherical solutes (methanol, water) while maintaining sufficient phase-space overlap rests on the alchemical integration along the bubble coordinate; however, the manuscript supplies no overlap metrics, lambda schedules, or variance estimates for the AIMD and MLMD runs, leaving open the possibility that the reported success is limited to well-behaved spherical cases.
Authors: We agree that explicit documentation of overlap and convergence would strengthen the presentation. The bubble coordinate was constructed precisely to ensure continuous and gradual solute-solvent decoupling for arbitrary shapes, avoiding the hard-core overlaps that produce singularities in standard alchemical paths. In the revised manuscript we now include the lambda schedules, the computed overlap integrals between adjacent windows, and the block-averaged variance estimates for both AIMD and MLMD trajectories. These quantities confirm adequate phase-space overlap for methanol and water, consistent with the method's design. The results for these non-spherical molecules therefore do not rely on special behavior limited to spherical solutes. revision: yes
-
Referee: [Ion Corrections subsection] For the ion corrections, the vacuum-water interface potential term is known to be sensitive to slab thickness, water-model details, and reference-potential choice; the text does not report convergence tests with respect to these parameters or demonstrate that the chosen value is independent of classical data, which directly undermines the assertion that the entire procedure is purely first-principles.
Authors: We acknowledge the known sensitivity of the interface potential. The value employed in the manuscript was obtained from separate first-principles DFT slab calculations performed with the same functional and basis set used in the production runs, without reference to classical force fields or experimental data. To address the referee's concern, the revised manuscript now reports explicit convergence tests with respect to slab thickness and the choice of reference potential within our ab initio framework. These tests show that the adopted correction remains stable under the conditions used for the Na+ calculations, thereby preserving the first-principles character of the overall procedure. revision: yes
-
Referee: [Results section] The validation is restricted to four small species; because the interface-potential correction and bubble construction must remain accurate when solute charge or shape changes, the limited test set does not yet establish the general applicability required by the abstract.
Authors: The current validation set is intentionally limited to four representative species (spherical and non-spherical, neutral and charged) to demonstrate feasibility across the method's intended scope. The bubble construction itself is formulated without reference to solute geometry, and the three periodic-boundary corrections for ions are derived from electrostatics that hold for any charge. We have added a dedicated paragraph in the revised manuscript discussing the expected transferability to larger solutes and different charge states, supported by the analytic form of the corrections. While broader benchmarking would be valuable, the existing results already substantiate the abstract's claims of applicability to arbitrary shapes and first-principles operation; we therefore do not view the test-set size as a fundamental limitation of the reported work. revision: partial
Circularity Check
No significant circularity: bubble method and ion corrections are independent constructions.
full rationale
The paper defines the bubble method explicitly as a coordinate transformation that avoids the end-state singularity by construction for arbitrary solute shapes, then applies standard literature corrections for periodic-boundary effects on ions (neutralizing background, image interactions, interface potential). These steps are presented as parameter-free first-principles protocols with no equations that reduce the final SFE values to fitted inputs, self-citations, or renamed empirical patterns. Validation on four species is external comparison, not part of the derivation chain itself. The central claim therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Molecular dynamics simulations provide sufficient sampling and ergodic exploration of phase space for the systems studied
- domain assumption Density functional theory approximations are adequate for the electronic structure of the tested molecules and ions
invented entities (1)
-
bubble method
no independent evidence
Reference graph
Works this paper leans on
-
[1]
and revPBE-D3 [25] exchange correlation functionals, in comparison with experimental values at T = 300 K and P = 1 bar. TABLE I. Solvation free energies of neutral molecules and Na+ obtained by the classical OPLS-AA force field, and PBE-D3 and revPBE-D3 exchange correlation functionals. Experimental values of the neutral molecules are from Ref. [26]. The ...
-
[2]
Given the symmetry of the curves aboutz= 25 ˚A, all data points are mapped to the left side for least squares fitting. We used a logistic sigmoid (or hyperbolic tangent) function, as employed by Kathmann [35], to perform the fitting. ˆϕ(z) = Vs 1 + e−β(z−z 0) +V 0 =V sSigm[β(z−z 0)] +V 0 = Vs 2 (1 + tanh[β(z−z 0) 2 ]) +V 0,(24) whereV 0 is the reference p...
-
[3]
Leung, K., Rempe, S. B. & von Lilienfeld, O. A. Ab initio molecular dynamics calculations of ion hydration free energies.The Journal of chemical physics130, 204507 (2009)
work page 2009
-
[4]
Luukkonen, S., Levesque, M., Belloni, L. & Borgis, D. Hydration free energies and solvation structures with molecular density functional theory in the hypernetted chain approximation. The Journal of Chemical Physics152, 064110 (2020)
work page 2020
-
[5]
Duarte Ramos Matos, G.et al.Approaches for calculating solvation free energies and en- thalpies demonstrated with an update of the freesolv database.Journal of Chemical & Engi- neering Data62, 1559–1569 (2017)
work page 2017
- [6]
-
[7]
Implicit solvent models in molecular dynamics simulations: A brief overview
Onufriev, A. Implicit solvent models in molecular dynamics simulations: A brief overview. Annual Reports in Computational Chemistry4, 125–137 (2008)
work page 2008
-
[8]
Decherchi, S., Masetti, M., Vyalov, I. & Rocchia, W. Implicit solvent methods for free energy estimation.European journal of medicinal chemistry91, 27–42 (2015)
work page 2015
-
[9]
Roux, B. & Simonson, T. Implicit solvent models.Biophysical chemistry78, 1–20 (1999)
work page 1999
- [10]
-
[11]
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. The Journal of chemical physics140, 084106 (2014)
work page 2014
-
[12]
Sinstein, M.et al.Efficient implicit solvation method for full potential dft.Journal of Chemical Theory and Computation13, 5582–5603 (2017)
work page 2017
-
[13]
Lu, N. & Kofke, D. A. Accuracy of free-energy perturbation calculations in molecular simu- lation. i. modeling.The Journal of Chemical Physics114, 7303–7311 (2001)
work page 2001
-
[14]
Shivakumar, D.et al.Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the opls force field.Journal of chemical theory and computation 6, 1509–1519 (2010)
work page 2010
-
[15]
Mezei, M. The finite difference thermodynamic integration, tested on calculating the hydration free energy difference between acetone and dimethylamine in water.The Journal of chemical physics86, 7084–7088 (1987)
work page 1987
-
[16]
Khanna, V., Monroe, J. I., Doherty, M. F. & Peters, B. Performing solvation free energy calculations in lammps using the decoupling approach.Journal of Computer-Aided Molecular Design34, 641–646 (2020)
work page 2020
-
[17]
Wang, F. & Cheng, J. Automated workflow for computation of redox potentials, acidity con- stants, and solvation free energies accelerated by machine learning.The Journal of Chemical Physics157, 024103 (2022). 18
work page 2022
-
[18]
Beutler, T. C., Mark, A. E., van Schaik, R. C., Gerber, P. R. & Van Gunsteren, W. F. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations.Chemical physics letters222, 529–539 (1994)
work page 1994
- [19]
-
[20]
Berendsen, H. J., van der Spoel, D. & van Drunen, R. Gromacs: A message-passing parallel molecular dynamics implementation.Computer physics communications91, 43–56 (1995)
work page 1995
-
[21]
Thompson, A. P.et al.Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales.Computer Physics Communications271, 108171 (2022)
work page 2022
-
[22]
Li, L., Totton, T. & Frenkel, D. Computational methodology for solubility prediction: Appli- cation to the sparingly soluble solutes.The Journal of chemical physics146, 214110 (2017)
work page 2017
-
[23]
Li, L., Totton, T. & Frenkel, D. Computational methodology for solubility prediction: Appli- cation to sparingly soluble organic/inorganic materials.The Journal of chemical physics149, 054102 (2018)
work page 2018
-
[24]
Lin, C., He, X., Xi, C., Zhang, Q. & Wang, L.-W. Ion solvation free energy calculations based on first-principles molecular dynamics thermodynamic integration.The Journal of Chemical Physics160, 184115 (2024)
work page 2024
-
[25]
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquids.Journal of the American Chemical Society118, 11225–11236 (1996)
work page 1996
-
[26]
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple (vol 77, pg 3865, 1996) (1997)
work page 1996
-
[27]
generalized gradient approximation made simple
Zhang, Y. & Yang, W. Comment on “generalized gradient approximation made simple”. Physical Review Letters80, 890 (1998). 19
work page 1998
-
[28]
Ben-Naim, A. & Marcus, Y. Solvation thermodynamics of nonionic solutes.The Journal of chemical physics81, 2016–2027 (1984)
work page 2016
-
[29]
Tissandier, M. D.et al.The proton’s absolute aqueous enthalpy and gibbs free energy of solvation from cluster-ion solvation data.The Journal of Physical Chemistry A102, 7787– 7794 (1998)
work page 1998
-
[30]
Flyvbjerg, H. & Petersen, H. G. Error estimates on averages of correlated data.The Journal of Chemical Physics91, 461–466 (1989)
work page 1989
-
[31]
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces.Physical review letters98, 146401 (2007)
work page 2007
-
[32]
Wang, H., Zhang, L., Han, J. & Weinan, E. Deepmd-kit: A deep learning package for many- body potential energy representation and molecular dynamics.Computer Physics Communi- cations228, 178–184 (2018)
work page 2018
-
[33]
Wang, R., Meraz, V. J. & Tiwary, P. Machine Learning Driven Advances in Molecular Dynamics of Bulk and Interfacial Aqueous Systems.Chemical Reviews(2026)
work page 2026
-
[34]
Schran, C., Brezina, K. & Marsalek, O. Committee neural network potentials control gen- eralization errors and enable active learning.The Journal of Chemical Physics153, 104105 (2020)
work page 2020
-
[35]
Kapil, V.et al.The first-principles phase diagram of monolayer nanoconfined water.Nature 609, 512–516 (2022)
work page 2022
-
[36]
H¨ unenberger, P. & Reif, M.Single-ion solvation: experimental and theoretical approaches to elusive thermodynamic quantities, vol. 3 (Royal Society of Chemistry, 2011)
work page 2011
-
[37]
Kathmann, S. M., Kuo, I.-F. W., Mundy, C. J. & Schenter, G. K. Understanding the surface potential of water.The Journal of Physical Chemistry B115, 4369–4377 (2011)
work page 2011
-
[38]
Smith, E., Bryk, T. & Haymet, A. D. Free energy of solvation of simple ions: Molecular- dynamics study of solvation of cl- and na+ in the ice/water interface.The Journal of chemical physics123, 034706 (2005). 20
work page 2005
-
[39]
Kelly, C. P., Cramer, C. J. & Truhlar, D. G. Aqueous solvation free energies of ions and ion- water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton.The Journal of Physical Chemistry B110, 16066–16081 (2006)
work page 2006
-
[40]
Toman´ ık, L., Muchov´ a, E. & Slav´ ıˇ cek, P. Solvation energies of ions with ensemble cluster- continuum approach.Physical Chemistry Chemical Physics22, 22357–22368 (2020)
work page 2020
-
[41]
Duignan, T. T., Baer, M. D., Schenter, G. K. & Mundy, C. J. Real single ion solvation free energies with quantum mechanical simulation.Chemical science8, 6131–6140 (2017)
work page 2017
-
[42]
Duignan, T. T., Baer, M. D., Schenter, G. K. & Mundy, C. J. Electrostatic solvation free energies of charged hard spheres using molecular dynamics with density functional theory interactions.The Journal of Chemical Physics147, 161716 (2017)
work page 2017
-
[43]
Freysoldt, C., Neugebauer, J. & Van de Walle, C. G. Fully ab initio finite-size corrections for charged-defect supercell calculations.Physics Review Letters102, 016402 (2009)
work page 2009
-
[44]
Harscher, A. & Lichte, H. Inelastic mean free path and mean inner potential of carbon foil and vitrified ice measured with electron holography.ICEM14, Cancun, Mexico31, 553–554 (1998)
work page 1998
-
[45]
Sellner, B. & Kathmann, S. M. A matter of quantum voltages.The Journal of Chemical Physics141, 18C534 (2014)
work page 2014
-
[46]
N.et al.Mean inner potential of liquid water.Physical Review Letters124, 065502 (2020)
Yesibolati, M. N.et al.Mean inner potential of liquid water.Physical Review Letters124, 065502 (2020)
work page 2020
-
[47]
In the CP2K code, the electrostatic potentialV(x, y, z) is stored on a 3D uniform real-space grid, with columns along thez-direction distributed across different MPI processes. While V(x, y, z) could in principle be written to a single file (e.g., a Gaussian cube file) for post- processing, this approach would require prohibitively large disk space. In ou...
-
[48]
Leung, K. Surface potential at the air- water interface computed using density functional theory.The Journal of Physical Chemistry Letters1, 496–499 (2010)
work page 2010
-
[49]
Cheng, J., Sulpizi, M. & Sprik, M. Redox potentials and p k a for benzoquinone from density functional theory based molecular dynamics.The Journal of chemical physics131, 154504 (2009)
work page 2009
-
[50]
Prozorov, T., Almeida, T. P., Kov´ acs, A. & Dunin-Borkowski, R. E. Off-axis electron holog- raphy of bacterial cells and magnetic nanoparticles in liquid.Journal of the Royal Society Interface14, 20170464 (2017)
work page 2017
-
[51]
Kusalik, P. G. & Svishchev, I. M. The spatial structure in liquid water.Science265, 1219– 1221 (1994)
work page 1994
-
[52]
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. Numerical integration of the cartesian equa- tions of motion of a system with constraints: molecular dynamics of n-alkanes.Journal of computational physics23, 327–341 (1977)
work page 1977
-
[53]
Evans, D. J. & Holian, B. L. The nose–hoover thermostat.The Journal of chemical physics 83, 4069–4074 (1985)
work page 1985
-
[54]
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions.Physical Review A31, 1695–1697 (1985)
work page 1985
-
[55]
E., Alejandre, J., L´ opez-Rend´ on, R., Jochim, A
Tuckerman, M. E., Alejandre, J., L´ opez-Rend´ on, R., Jochim, A. L. & Martyna, G. J. A liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble.Journal of Physics A: Mathematical and General39, 5629 (2006)
work page 2006
-
[56]
K¨ uhne, T. D.et al.CP2K: An electronic structure and molecular dynamics software package- Quickstep: Efficient and accurate electronic structure calculations.The Journal of Chemical Physics152, 194103 (2020)
work page 2020
-
[57]
VandeVondele, J.et al.Quickstep: Fast and accurate density functional calculations using a mixed gaussian and plane waves approach.Computer Physics Communications167, 103–128 22 (2005)
work page 2005
-
[58]
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space gaussian pseudopotentials.Phys- ical Review B54, 1703 (1996)
work page 1996
-
[59]
Hartwigsen, C., Gœdecker, S. & Hutter, J. Relativistic separable dual-space gaussian pseu- dopotentials from h to rn.Physical Review B58, 3641 (1998)
work page 1998
-
[60]
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu. The Journal of chemical physics132, 154104 (2010)
work page 2010
-
[61]
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling.The Journal of chemical physics126, 014101 (2007)
work page 2007
-
[62]
Martyna, G. J., Tobias, D. J. & Klein, M. L. Constant pressure molecular dynamics algorithms. The Journal of chemical physics101, 4177–4189 (1994)
work page 1994
-
[63]
Guidon, M., Hutter, J. & VandeVondele, J. Auxiliary density matrix methods for hartree- fock exchange calculations.Journal of chemical theory and computation6, 2348–2364 (2010). 23 (a) (b) (c) (d) Expanding Switching FIG. 1. Schematic illustration of the bubble method scheme. (a) In the absence of solute-solvent interactions, a spherical bubble forms and ...
work page 2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.