Recognition: unknown
Learning biophysical models of gene regulation with probability flow matching
Pith reviewed 2026-05-07 16:49 UTC · model grok-4.3
The pith
Probability Flow Matching learns biophysically consistent stochastic models of gene regulation from single-cell data
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
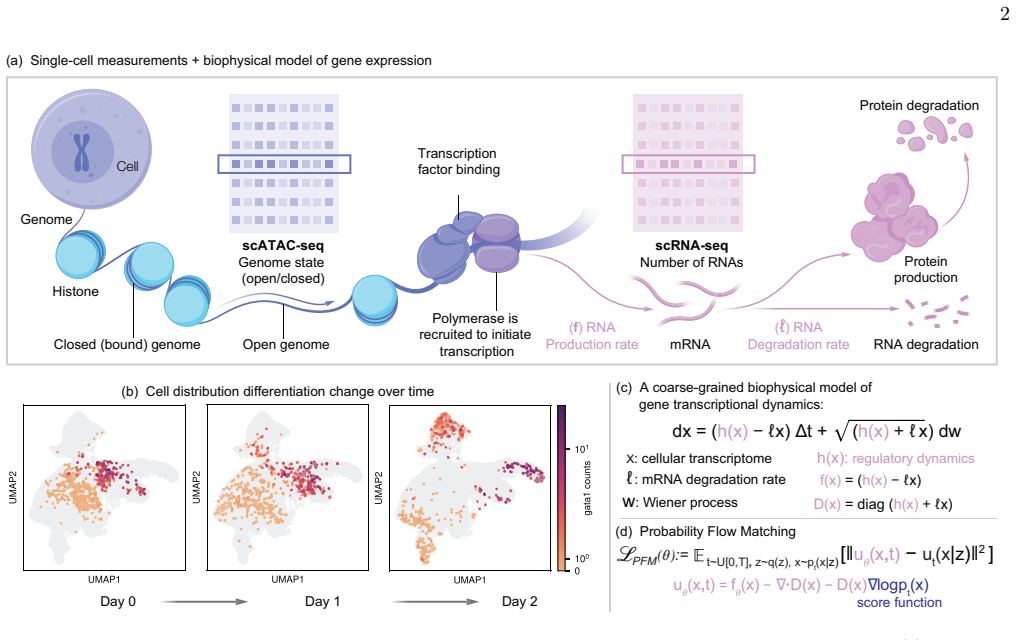
Models with similar interpolation accuracy can encode fundamentally different dynamics, and only biophysically consistent formulations accurately capture mechanisms of lineage transitions, fate specification, and gene perturbation responses in hematopoiesis datasets.
What carries the argument
Probability Flow Matching, a framework for learning biophysically consistent stochastic processes directly from time-resolved single-cell measurements that distinguishes interpolating models from mechanistically faithful ones.
If this is right
- Biophysically constrained models enable accurate prediction of cellular responses to gene perturbations not seen during training.
- The framework supports simultaneous inference of regulatory dynamics together with proliferation and death rates from unbalanced populations.
- Mechanistic interpretability becomes possible without sacrificing the ability to fit high-dimensional single-cell time courses.
- Generalization beyond the training data improves when physical constraints replace pure statistical fitting.
Where Pith is reading between the lines
- The approach could be tested on other differentiation systems such as neural or immune cell development to check whether biophysical consistency remains necessary.
- Learned models might serve as simulators for screening potential gene interventions before experimental validation.
- Purely data-driven methods on single-cell data risk producing dynamics that fit observations but lack predictive power for biological mechanisms.
Load-bearing premise
Enforcing biophysical consistency in the learned stochastic processes is feasible from time-resolved single-cell data and sufficient to distinguish models that capture true biological mechanisms from those that merely interpolate transcriptomes.
What would settle it
An experiment showing that a non-biophysically constrained model accurately predicts held-out lineage transitions, fate specifications, and perturbation responses would falsify the necessity of biophysical consistency.
Figures




read the original abstract
Cellular differentiation is governed by gene regulatory networks, the high-dimensional stochastic biochemical systems that determine the transcriptional landscape and mediate cellular responses to signals and perturbations. Although single-cell RNA sequencing provides quantitative snapshots of the transcriptome, current methods for inferring gene-regulatory dynamics often lack mechanistic interpretability and fail to generalize to unseen conditions. Here we introduce Probability Flow Matching (PFM), a scalable framework for learning biophysically consistent stochastic processes directly from time-resolved single-cell measurements. Applying PFM to three hematopoiesis datasets, we show that models with similar interpolation accuracy can encode fundamentally different dynamics, with only biophysically consistent formulations accurately capturing mechanisms of lineage transitions, fate specification, and gene perturbation responses. We further demonstrate that PFM accommodates unbalanced populations, enabling simultaneous inference of cellular proliferation and death dynamics. Together, these results establish PFM as a flexible, scalable framework for integrating mechanistic modeling with single-cell omics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces Probability Flow Matching (PFM), a scalable framework for learning biophysically consistent stochastic processes that model gene regulatory dynamics directly from time-resolved single-cell RNA-seq data. Applied to three hematopoiesis datasets, it claims that models achieving similar interpolation accuracy can encode different dynamics, but only biophysically consistent formulations (respecting constraints such as non-negative rates and mass-action structure) accurately recover mechanisms of lineage transitions, fate specification, and gene perturbation responses. The method also accommodates unbalanced populations by jointly inferring proliferation and death rates.
Significance. If validated, PFM would offer a principled way to embed mechanistic biophysical constraints into high-dimensional single-cell trajectory inference, addressing the common failure of purely statistical models to generalize to perturbations or to yield interpretable regulatory mechanisms. The ability to handle unbalanced populations is a practical strength for real datasets.
major comments (3)
- [Results (hematopoiesis datasets)] Results section (hematopoiesis applications): the central claim that biophysical consistency selects models that 'accurately capture mechanisms' is supported only by agreement with known markers and held-out perturbations on real data. No synthetic benchmarks with known ground-truth GRN vector fields are presented, so it remains unclear whether the constrained PFM recovers the true drift/diffusion better than an unconstrained model that merely matches observed marginals.
- [Methods] Methods (PFM formulation and biophysical constraints): the paper does not provide explicit details on how biophysical consistency is enforced (e.g., via hard constraints, penalties, or architecture choices) nor quantitative verification that the learned processes satisfy the claimed properties (non-negativity, mass-action structure) after training. This makes it difficult to assess whether the reported superiority is due to the constraints or to other modeling choices.
- [Results] Validation metrics: across the three datasets, the manuscript supplies no error bars, cross-validation details, or data-exclusion criteria for the interpolation and perturbation experiments. Without these, the claim that 'models with similar interpolation accuracy' differ in mechanistic fidelity cannot be rigorously evaluated.
minor comments (2)
- [Methods] Notation for the probability flow and the stochastic process should be introduced with a clear equation reference early in the Methods section to aid readability.
- [Figures] Figure legends for the hematopoiesis visualizations should explicitly state which panels correspond to constrained vs. unconstrained PFM runs.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback on our manuscript. We address each major comment below and indicate the revisions we will incorporate to strengthen the presentation and rigor of the work.
read point-by-point responses
-
Referee: Results section (hematopoiesis applications): the central claim that biophysical consistency selects models that 'accurately capture mechanisms' is supported only by agreement with known markers and held-out perturbations on real data. No synthetic benchmarks with known ground-truth GRN vector fields are presented, so it remains unclear whether the constrained PFM recovers the true drift/diffusion better than an unconstrained model that merely matches observed marginals.
Authors: We agree that synthetic benchmarks with known ground-truth vector fields would provide direct evidence of dynamics recovery. However, generating high-dimensional synthetic single-cell data that faithfully reproduces the marginal distributions, noise characteristics, and lineage structure of real hematopoiesis datasets while supplying exact ground-truth GRN vector fields is technically challenging and not straightforward. Our validation strategy instead leverages agreement with established biological markers and performance on held-out perturbation experiments as biologically meaningful proxies. We will add a dedicated paragraph in the Discussion section explaining this choice and the associated limitations. revision: partial
-
Referee: Methods (PFM formulation and biophysical constraints): the paper does not provide explicit details on how biophysical consistency is enforced (e.g., via hard constraints, penalties, or architecture choices) nor quantitative verification that the learned processes satisfy the claimed properties (non-negativity, mass-action structure) after training. This makes it difficult to assess whether the reported superiority is due to the constraints or to other modeling choices.
Authors: We appreciate this observation. Biophysical consistency is enforced via a combination of architectural parameterization (non-negative outputs for reaction rates via softplus activations) and auxiliary penalty terms in the objective that penalize deviations from mass-action structure. We will expand the Methods section with the precise mathematical formulations of these mechanisms and add post-training verification metrics, including the percentage of negative rates observed across models and quantitative measures of mass-action adherence on held-out data. revision: yes
-
Referee: Validation metrics: across the three datasets, the manuscript supplies no error bars, cross-validation details, or data-exclusion criteria for the interpolation and perturbation experiments. Without these, the claim that 'models with similar interpolation accuracy' differ in mechanistic fidelity cannot be rigorously evaluated.
Authors: We agree that these details are necessary for rigorous evaluation. In the revised manuscript we will report error bars as standard deviations over five independent training runs for all interpolation and perturbation metrics. We will also add explicit descriptions of the cross-validation procedure (held-out time points and cells) and the data-exclusion criteria applied during preprocessing of the three hematopoiesis datasets. revision: yes
Circularity Check
No significant circularity; biophysical constraints imposed externally and validated against external biological benchmarks
full rationale
The paper introduces Probability Flow Matching (PFM) as a new framework for learning stochastic processes from time-resolved single-cell data, with biophysical consistency (e.g., non-negative rates, mass-action structure) imposed as an external modeling choice rather than derived from the target results. The central claim—that only biophysically consistent models capture true mechanisms—is tested via agreement with known hematopoiesis lineage markers and held-out perturbation responses, which constitute independent external benchmarks. No self-definitional reductions, fitted inputs renamed as predictions, or load-bearing self-citations appear in the derivation chain. The method remains self-contained against these external references.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Biophysical consistency can be defined and enforced as constraints on the learned stochastic processes
Reference graph
Works this paper leans on
-
[1]
Transition states and cell fate decisions in epigenetic land- scapes.Nature Reviews Genetics, 17(11):693–703, 2016
Naomi Moris, Cristina Pina, and Alfonso Martinez Arias. Transition states and cell fate decisions in epigenetic land- scapes.Nature Reviews Genetics, 17(11):693–703, 2016
2016
-
[2]
Defining cell types and states with single- cell genomics.Genome research, 25(10):1491–1498, 2015
Cole Trapnell. Defining cell types and states with single- cell genomics.Genome research, 25(10):1491–1498, 2015
2015
-
[3]
Human haematopoietic stem cell lineage commitment is a continuous process.Nature cell biology, 19(4):271–281, 2017
Lars Velten, Simon F Haas, Simon Raffel, Sandra Blaszkiewicz, Saiful Islam, Bianca P Hennig, Christoph Hirche, Christoph Lutz, Eike C Buss, Daniel Nowak, et al. Human haematopoietic stem cell lineage commitment is a continuous process.Nature cell biology, 19(4):271–281, 2017
2017
-
[4]
Population snapshots predict early haematopoi- etic and erythroid hierarchies.Nature, 555(7694):54–60, 2018
Betsabeh Khoramian Tusi, Samuel L Wolock, Caleb Wein- reb, Yung Hwang, Daniel Hidalgo, Rapolas Zilionis, Ari Waisman, Jun R Huh, Allon M Klein, and Merav So- colovsky. Population snapshots predict early haematopoi- etic and erythroid hierarchies.Nature, 555(7694):54–60, 2018
2018
-
[5]
Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets.Cell, 161(5):1202– 1214, 2015
Evan Z Macosko, Anindita Basu, Rahul Satija, James Nemesh, Karthik Shekhar, Melissa Goldman, Itay Tirosh, Allison R Bialas, Nolan Kamitaki, Emily M Martersteck, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets.Cell, 161(5):1202– 1214, 2015
2015
-
[6]
Single-cell chromatin accessibility reveals principles of regulatory variation.Nature, 523(7561):486–490, 2015
Jason D Buenrostro, Bryan Wu, Uli M Litzenburger, Darin Ruff, Michael L Gonzales, Matthew P Snyder, Howard Y Chang, and William J Greenleaf. Single-cell chromatin accessibility reveals principles of regulatory variation.Nature, 523(7561):486–490, 2015
2015
-
[7]
Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells.Science, 362(6413):eaau1783, 2018
Bogdan Bintu, Laura J Mateo, Jonathan H Su, Nicholas A Sinnott-Armstrong, Marissa Parker, Shaul S Kinrot, Ken Yamaya, Alistair N Boettiger, and Xiaowei Zhuang. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells.Science, 362(6413):eaau1783, 2018
2018
-
[8]
From haematopoi- etic stem cells to complex differentiation landscapes.Na- ture, 553(7689):418–426, 2018
Elisa Laurenti and Berthold Göttgens. From haematopoi- etic stem cells to complex differentiation landscapes.Na- ture, 553(7689):418–426, 2018
2018
-
[9]
Deciphering cell fate decision by integrated single-cell sequencing analysis.Annual review of biomedical data science, 3(1):1–22, 2020
Sagar and Dominic Grün. Deciphering cell fate decision by integrated single-cell sequencing analysis.Annual review of biomedical data science, 3(1):1–22, 2020
2020
-
[10]
Joint probabilistic modeling of single-cell multi-omic data with totalvi.Nature methods, 18(3):272–282, 2021
Adam Gayoso, Zoë Steier, Romain Lopez, Jeffrey Regier, Kristopher L Nazor, Aaron Streets, and Nir Yosef. Joint probabilistic modeling of single-cell multi-omic data with totalvi.Nature methods, 18(3):272–282, 2021
2021
-
[11]
A roadmap for multi-omics data integration using deep learn- ing.Briefings in Bioinformatics, 23(1):bbab454, 2022
Mingon Kang, Euiseong Ko, and Tesfaye B Mersha. A roadmap for multi-omics data integration using deep learn- ing.Briefings in Bioinformatics, 23(1):bbab454, 2022
2022
-
[12]
Machine learning for multi-omics data integration in cancer.Iscience, 25(2), 2022
Zhaoxiang Cai, Rebecca C Poulos, Jia Liu, and Qing Zhong. Machine learning for multi-omics data integration in cancer.Iscience, 25(2), 2022
2022
-
[13]
Lineage tracing on tran- scriptional landscapes links state to fate during differenti- ation.Science, 367(6479):eaaw3381, 2020
Caleb Weinreb, Alejo Rodriguez-Fraticelli, Fernando D Camargo, and Allon M Klein. Lineage tracing on tran- scriptional landscapes links state to fate during differenti- ation.Science, 367(6479):eaaw3381, 2020
2020
-
[14]
Deep generative modeling for single-cell transcriptomics.Nature methods, 15(12):1053– 1058, 2018
Romain Lopez, Jeffrey Regier, Michael B Cole, Michael I Jordan, and Nir Yosef. Deep generative modeling for single-cell transcriptomics.Nature methods, 15(12):1053– 1058, 2018
2018
-
[15]
Dimensionality re- duction for visualizing single-cell data using umap.Nature biotechnology, 37(1):38–44, 2019
Etienne Becht, Leland McInnes, John Healy, Charles- Antoine Dutertre, Immanuel WH Kwok, Lai Guan Ng, Florent Ginhoux, and Evan W Newell. Dimensionality re- duction for visualizing single-cell data using umap.Nature biotechnology, 37(1):38–44, 2019
2019
-
[16]
Fundamental limits on dynamic inference from single-cell snapshots.Proceed- ings of the National Academy of Sciences, 117(2):775–785, 2020
Caleb Weinreb, Samuel Wolock, Brian K Tusi, Merav Socolovsky, and Allon M Klein. Fundamental limits on dynamic inference from single-cell snapshots.Proceed- ings of the National Academy of Sciences, 117(2):775–785, 2020
2020
-
[17]
The dynamics and regulators of cell fate decisions are revealedbypseudotemporalorderingofsinglecells.Nature biotechnology, 32(4):381–386, 2014
Cole Trapnell, Davide Cacchiarelli, Jonna Grimsby, Prapti Pokharel, Shuqiang Li, Michael Morse, Niall J Lennon, Kenneth J Livak, Tarjei S Mikkelsen, and John L Rinn. The dynamics and regulators of cell fate decisions are revealedbypseudotemporalorderingofsinglecells.Nature biotechnology, 32(4):381–386, 2014
2014
-
[18]
Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics.BMC genomics, 19(1):477, 2018
Kelly Street, Davide Risso, Russell B Fletcher, Diya Das, John Ngai, Nir Yosef, Elizabeth Purdom, and Sandrine Dudoit. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics.BMC genomics, 19(1):477, 2018
2018
-
[19]
Visualizing structure and transitions in high-dimensional biological data.Nature biotechnology, 37(12):1482–1492, 2019
Kevin R Moon, David Van Dijk, Zheng Wang, Scott Gigante, Daniel B Burkhardt, William S Chen, Kristina Yim, Antonia van den Elzen, Matthew J Hirn, Ronald R Coifman, et al. Visualizing structure and transitions in high-dimensional biological data.Nature biotechnology, 37(12):1482–1492, 2019
2019
-
[20]
Wishbone identifies bifurcating developmental trajectories from single-cell data.Nature biotechnology, 34(6):637–645, 2016
Manu Setty, Michal D Tadmor, Shlomit Reich-Zeliger, Ori Angel, Tomer M Salame, Pooja Kathail, Karthik Choi, Sean C Bendall, Nir Friedman, and Dana Pe’er. Wishbone identifies bifurcating developmental trajectories from single-cell data.Nature biotechnology, 34(6):637–645, 2016
2016
-
[21]
A comparison of single-cell trajectory inference methods.Nature biotechnology, 37(5):547–554, 2019
Wouter Saelens, Robrecht Cannoodt, Helena Todorov, and Yvan Saeys. A comparison of single-cell trajectory inference methods.Nature biotechnology, 37(5):547–554, 2019
2019
-
[22]
Eleven grand challenges in single-cell data science.Genome biology, 21(1):31, 2020
David Lähnemann, Johannes Köster, Ewa Szczurek, Davis J McCarthy, Stephanie C Hicks, Mark D Robinson, Catalina A Vallejos, Kieran R Campbell, Niko Beeren- winkel, Ahmed Mahfouz, et al. Eleven grand challenges in single-cell data science.Genome biology, 21(1):31, 2020
2020
-
[23]
Combined mechanistic modeling and machine-learning approaches in systems biology-a systematic literature re- view.Comput
Anna Procopio, Giuseppe Cesarelli, Leandro Donisi, Alessio Merola, Francesco Amato, Carlo Cosentino, et al. Combined mechanistic modeling and machine-learning approaches in systems biology-a systematic literature re- view.Comput. Methods Programs Biomed., 240:107681, 2023
2023
-
[24]
Rna velocity of single cells.Nature, 560(7719):494–498, 2018
Gioele La Manno, Ruslan Soldatov, Amit Zeisel, Emelie Braun, Hannah Hochgerner, Viktor Petukhov, Katja Lid- schreiber, Maria E Kastriti, Peter Lönnerberg, Alessan- dro Furlan, et al. Rna velocity of single cells.Nature, 560(7719):494–498, 2018
2018
-
[25]
Iden- tification of gene regulation models from single-cell data
Lisa Weber, William Raymond, and Brian Munsky. Iden- tification of gene regulation models from single-cell data. Physical biology, 15(5):055001, 2018
2018
-
[26]
Inferring gene regulatory networks from single-cell data: a mechanistic approach.BMC systems biology, 11(1):105, 2017
Ulysse Herbach, Arnaud Bonnaffoux, Thibault Espinasse, and Olivier Gandrillon. Inferring gene regulatory networks from single-cell data: a mechanistic approach.BMC systems biology, 11(1):105, 2017
2017
-
[27]
Geoffrey Schiebinger, Jian Shu, Marcin Tabaka, Brian Cleary, Vidya Subramanian, Aryeh Solomon, Joshua Gould, Siyan Liu, Stacie Lin, Peter Berube, Lia Lee, Jenny Chen, Justin Brumbaugh, Philippe Rigollet, Kon- 13 radHochedlinger, RudolfJaenisch, AvivRegev, andEricS. Lander. Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental ...
2019
-
[28]
Reversed graph embedding resolves complex single-cell trajectories
Xiaojie Qiu, Qian Mao, Yajing Tang, Lin Wang, Rajiv Chawla, Hannah A Pliner, and Cole Trapnell. Reversed graph embedding resolves complex single-cell trajectories. Nature methods, 14(10):979–982, 2017
2017
-
[29]
Learning stochastic processes with intrinsic noise from cross-sectional biological data.Proceedings of the National Academy of Sciences, 122(37):e2420621122, 2025
Suryanarayana Maddu, Victor Chardès, and Michael J Shelley. Learning stochastic processes with intrinsic noise from cross-sectional biological data.Proceedings of the National Academy of Sciences, 122(37):e2420621122, 2025
2025
-
[30]
Scalable unbalanced optimal transport using generative adversarial networks
Karren D Yang and Caroline Uhler. Scalable unbalanced optimal transport using generative adversarial networks. arXiv preprint arXiv:1810.11447, 2018
-
[31]
Reconstructing developmental land- scapes and trajectories from single-cell data.Current Opinion in Systems Biology, 27:100351, September 2021
Geoffrey Schiebinger. Reconstructing developmental land- scapes and trajectories from single-cell data.Current Opinion in Systems Biology, 27:100351, September 2021
2021
-
[32]
Optimal transport analysis reveals trajectories in steady-state sys- tems.PLOS Computational Biology, 17(12):e1009466, December 2021
Stephen Zhang, Anton Afanassiev, Laura Greenstreet, Tetsuya Matsumoto, and Geoffrey Schiebinger. Optimal transport analysis reveals trajectories in steady-state sys- tems.PLOS Computational Biology, 17(12):e1009466, December 2021
2021
-
[33]
Stark, Gabriele Gut, Ja- cobo Sarabia del Castillo, Mitch Levesque, Kjong-Van Lehmann, Lucas Pelkmans, Andreas Krause, and Gunnar Rätsch
Charlotte Bunne, Stefan G. Stark, Gabriele Gut, Ja- cobo Sarabia del Castillo, Mitch Levesque, Kjong-Van Lehmann, Lucas Pelkmans, Andreas Krause, and Gunnar Rätsch. Learning single-cell perturbation responses using neural optimal transport.Nature Methods, pages 1–10, September 2023
2023
-
[34]
Trajectorynet: A dynamic optimal transport network for modeling cellular dynamics
Alexander Tong, Jessie Huang, Guy Wolf, David Van Dijk, and Smita Krishnaswamy. Trajectorynet: A dynamic optimal transport network for modeling cellular dynamics. InInternational conference on machine learning, pages 9526–9536. PMLR, 2020
2020
-
[35]
Trajectory Inference via Mean-field Langevin in Path Space.Advances in Neural Information Processing Systems, 35:16731–16742, December 2022
Lénaïc Chizat, Stephen Zhang, Matthieu Heitz, and Ge- offrey Schiebinger. Trajectory Inference via Mean-field Langevin in Path Space.Advances in Neural Information Processing Systems, 35:16731–16742, December 2022
2022
-
[36]
Towards a mathematical theory of trajectory inference, April 2023
Hugo Lavenant, Stephen Zhang, Young-Heon Kim, and Geoffrey Schiebinger. Towards a mathematical theory of trajectory inference, April 2023
2023
-
[37]
The Schrödinger Bridge between Gaussian Measures has a Closed Form
Charlotte Bunne, Ya-Ping Hsieh, Marco Cuturi, and An- dreas Krause. The Schrödinger Bridge between Gaussian Measures has a Closed Form. InProceedings of The 26th International Conference on Artificial Intelligence and Statistics, pages 5802–5833. PMLR, April 2023
2023
-
[38]
Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data.Bioin- formatics, 31(12):i89–i96, 2015
Andrea Ocone, Laleh Haghverdi, Nikola S Mueller, and Fabian J Theis. Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data.Bioin- formatics, 31(12):i89–i96, 2015
2015
-
[39]
Scode: an efficient regula- tory network inference algorithm from single-cell rna-seq during differentiation.Bioinformatics, 33(15):2314–2321, 2017
Hirotaka Matsumoto, Hisanori Kiryu, Chikara Furusawa, Minoru SH Ko, Shigeru BH Ko, Norio Gouda, Tetsutaro Hayashi, and Itoshi Nikaido. Scode: an efficient regula- tory network inference algorithm from single-cell rna-seq during differentiation.Bioinformatics, 33(15):2314–2321, 2017
2017
-
[40]
A bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data.Bioinformatics, 34(6):964–970, 2018
Manuel Sanchez-Castillo, David Blanco, Isabel M Tienda- Luna, MC Carrion, and Yufei Huang. A bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data.Bioinformatics, 34(6):964–970, 2018
2018
-
[41]
Learning Population-Level Diffusions with Generative RNNs
Tatsunori Hashimoto, David Gifford, and Tommi Jaakkola. Learning Population-Level Diffusions with Generative RNNs. InProceedings of The 33rd Interna- tional Conference on Machine Learning, pages 2417–2426. PMLR, June 2016
2016
-
[42]
Generative modeling of single-cell time series with prescient enables prediction of cell trajectories with interventions.Nature communications, 12(1):3222, 2021
Grace Hui Ting Yeo, Sachit D Saksena, and David K Gifford. Generative modeling of single-cell time series with prescient enables prediction of cell trajectories with interventions.Nature communications, 12(1):3222, 2021
2021
-
[43]
Flow matching for gen- erative modeling
Yotam Lipman, Zhoutong Jiang, Joshua B Tenenbaum, William T Freeman, Roger Grosse, Danilo Jimenez Rezende, and Taylor Webber. Flow matching for gen- erative modeling. InAdvances in Neural Information Processing Systems, volume 35, 2022
2022
-
[44]
Yaron Lipman, Marton Havasi, Peter Holderrieth, Neta Shaul, Matt Le, Brian Karrer, Ricky TQ Chen, David Lopez-Paz, Heli Ben-Hamu, and Itai Gat. Flow matching guide and code.arXiv preprint arXiv:2412.06264, 2024
work page internal anchor Pith review arXiv 2024
-
[45]
Simulation-free schrödinger bridges via score and flow matching
Alexander Y Tong, Nikolay Malkin, Kilian Fatras, Lazar Atanackovic, Yanlei Zhang, Guillaume Huguet, Guy Wolf, and Yoshua Bengio. Simulation-free schrödinger bridges via score and flow matching. InInternational Conference on Artificial Intelligence and Statistics, pages 1279–1287. PMLR, 2024
2024
-
[46]
Modeling complex system dynamics with flow matching across time and conditions
Martin Rohbeck, Edward De Brouwer, Charlotte Bunne, Jan-Christian Huetter, Anne Biton, Kelvin Y Chen, Aviv Regev, and Romain Lopez. Modeling complex system dynamics with flow matching across time and conditions. InThe Thirteenth International Conference on Learning Representations, 2025
2025
-
[47]
Stochastic force inference via density estimation
Victor Chardès, Suryanarayana Maddu, and Michael J Shelley. Stochastic force inference via density estimation. InNeurIPS 2023 AI for Science Workshop
2023
-
[48]
Inferring stochastic dynamics with growth from cross-sectional data
Stephen Y Zhang, Suryanarayana Maddu, Xiaojie Qiu, and Victor Chardès. Inferring stochastic dynamics with growth from cross-sectional data. InThe Thirty-ninth Annual Conference on Neural Information Processing Sys- tems, 2025
2025
-
[49]
Zheng, Genevieve Stein-O’Brien, Leandros Boukas, Loyal A
Shijie C. Zheng, Genevieve Stein-O’Brien, Leandros Boukas, Loyal A. Goff, and Kasper D. Hansen. Pump- ing the brakes on RNA velocity by understanding and interpreting RNA velocity estimates.Genome Biology, 24(1):246, October 2023
2023
-
[50]
Royer, Alejandro Granados, and Merlin Lange
Sarah Ancheta, Leah Dorman, Guillaume Le Treut, Abel Gurung, Loïc A. Royer, Alejandro Granados, and Merlin Lange. Challenges and Progress in RNA Velocity: Com- parative Analysis Across Multiple Biological Contexts, June 2024
2024
-
[51]
Generalizing rna velocity to transient cell states through dynamical modeling.Nature biotechnology, 38(12):1408–1414, 2020
Volker Bergen, Marius Lange, Stefan Peidli, F Alexander Wolf, and Fabian J Theis. Generalizing rna velocity to transient cell states through dynamical modeling.Nature biotechnology, 38(12):1408–1414, 2020
2020
-
[52]
Jalihal, Jeffrey N
Aditya Pratapa, Amogh P. Jalihal, Jeffrey N. Law, Aditya Bharadwaj, and T. M. Murali. Benchmarking algorithms for gene regulatory network inference from single-cell tran- scriptomic data.Nature Methods, 17(2):147–154, February 2020
2020
-
[53]
Generative models of cell dynamics: from neural odes to flow matching.Communications Biology, 2026
Till Richter, Weixu Wang, Alessandro Palma, and Fabian J Theis. Generative models of cell dynamics: from neural odes to flow matching.Communications Biology, 2026
2026
-
[54]
Score-Based Generative Modeling through Stochastic Differential Equations
Yang Song, Jascha Sohl-Dickstein, Diederik P Kingma, Abhishek Kumar, Stefano Ermon, and Ben Poole. Score- based generative modeling through stochastic differential equations.arXiv preprint arXiv:2011.13456, 2020
work page internal anchor Pith review arXiv 2011
-
[55]
Score-based generative modeling through stochastic differential equations
Yang Song and Stefano Ermon. Score-based generative modeling through stochastic differential equations. In 14 International Conference on Learning Representations, 2021
2021
-
[56]
Neural ordinary differential equations
Ricky TQ Chen, Yulia Rubanova, Jesse Bettencourt, and David K Duvenaud. Neural ordinary differential equations. InAdvances in neural information processing systems, volume 31. Curran Associates, Inc., 2018
2018
-
[57]
Discrete regula- tory modules instruct hematopoietic lineage commitment and differentiation.Nature Communications, 12(1):6790, 2021
Grigorios Georgolopoulos, Nikoletta Psatha, Mineo Iwata, AndrewNishida, TannishthaSom, MinasYiangou, JohnA Stamatoyannopoulos, and Jeff Vierstra. Discrete regula- tory modules instruct hematopoietic lineage commitment and differentiation.Nature Communications, 12(1):6790, 2021
2021
-
[58]
From hi-c contact map to three-dimensional organization of interphase human chromosomes.Physical Review X, 11(1):011051, 2021
Guang Shi and D Thirumalai. From hi-c contact map to three-dimensional organization of interphase human chromosomes.Physical Review X, 11(1):011051, 2021
2021
-
[59]
Digit patterning is controlled by a bmp- sox9-wnt turing network modulated by morphogen gradi- ents.Science, 345(6196):566–570, 2014
Jelena Raspopovic, Luciano Marcon, Laura Russo, and James Sharpe. Digit patterning is controlled by a bmp- sox9-wnt turing network modulated by morphogen gradi- ents.Science, 345(6196):566–570, 2014
2014
-
[60]
Spatiotemporal modeling of molecular holograms.Cell, 187(26):7351–7373, 2024
Xiaojie Qiu, Daniel Y Zhu, Yifan Lu, Jiajun Yao, Zehua Jing, Kyung Hoi Min, Mengnan Cheng, Hailin Pan, Lulu Zuo, Samuel King, et al. Spatiotemporal modeling of molecular holograms.Cell, 187(26):7351–7373, 2024
2024
-
[61]
Estimation of non- normalized statistical models by score matching.Journal of Machine Learning Research, 6(4), 2005
Aapo Hyvärinen and Peter Dayan. Estimation of non- normalized statistical models by score matching.Journal of Machine Learning Research, 6(4), 2005
2005
-
[62]
Re- constructing growth and dynamic trajectories from single- cell transcriptomics data.Nature Machine Intelligence, 6(1):25–39, 2024
Yutong Sha, Yuchi Qiu, Peijie Zhou, and Qing Nie. Re- constructing growth and dynamic trajectories from single- cell transcriptomics data.Nature Machine Intelligence, 6(1):25–39, 2024
2024
-
[63]
Sliced score matching: A scalable approach to density and score estimation
Yang Song, Sahaj Garg, Jiaxin Shi, and Stefano Ermon. Sliced score matching: A scalable approach to density and score estimation. InUncertainty in artificial intelligence, pages 574–584. PMLR, 2020
2020
-
[64]
Hematopoietic differentiation is characterized by a transient peak of entropy at a single-cell level.BMC biology, 20(1):60, 2022
Charles Dussiau, Agathe Boussaroque, Mathilde Gaillard, Clotilde Bravetti, Laila Zaroili, Camille Knosp, Chloé Friedrich, Philippe Asquier, Lise Willems, Laurent Quint, et al. Hematopoietic differentiation is characterized by a transient peak of entropy at a single-cell level.BMC biology, 20(1):60, 2022
2022
-
[65]
Combinatorial function of transcription factors and cofactors.Current opinion in genetics & development, 43:73–81, 2017
Franziska Reiter, Sebastian Wienerroither, and Alexander Stark. Combinatorial function of transcription factors and cofactors.Current opinion in genetics & development, 43:73–81, 2017
2017
-
[66]
Abso- lute quantification of transcription factors reveals princi- ples of gene regulation in erythropoiesis.Molecular cell, 78(5):960–974, 2020
Mark A Gillespie, Carmen G Palii, Daniel Sanchez- Taltavull, Paul Shannon, William JR Longabaugh, Damien J Downes, Karthi Sivaraman, Herbert M Es- pinoza, Jim R Hughes, Nathan D Price, et al. Abso- lute quantification of transcription factors reveals princi- ples of gene regulation in erythropoiesis.Molecular cell, 78(5):960–974, 2020
2020
-
[67]
Miller, Daniel B
Benjamin DeMeo, Charlotte Nesbitt, Samuel A. Miller, Daniel B. Burkhardt, Inna Lipchina, Doris Fu, Peter Holderrieth, David Kim, Sergey Kolchenko, Artur Sza- lata, Ishan Gupta, Christine Kerr, Thomas Pfefer, Ra- ziel Rojas-Rodriguez, Sunil Kuppassani, Laurens Kruide- nier, Parul B. Doshi, Mahdi Zamanighomi, James J. Collins, Alex K. Shalek, Fabian J. Thei...
2025
-
[68]
Noise distorts the epigenetic landscape and shapes cell- fate decisions.Cell Systems, 13(1):83–102, 2022
Megan A Coomer, Lucy Ham, and Michael PH Stumpf. Noise distorts the epigenetic landscape and shapes cell- fate decisions.Cell Systems, 13(1):83–102, 2022
2022
-
[69]
Inverse dirichlet weight- ing enables reliable training of physics informed neural networks.Machine Learning: Science and Technology, 3(1):015026, 2022
Suryanarayana Maddu, Dominik Sturm, Christian L Müller, and Ivo F Sbalzarini. Inverse dirichlet weight- ing enables reliable training of physics informed neural networks.Machine Learning: Science and Technology, 3(1):015026, 2022
2022
-
[70]
Stochasticity and cell fate.science, 320(5872):65–68, 2008
Richard Losick and Claude Desplan. Stochasticity and cell fate.science, 320(5872):65–68, 2008
2008
-
[71]
Regulated noise in the epigenetic landscape of development and disease
Elisabet Pujadas and Andrew P Feinberg. Regulated noise in the epigenetic landscape of development and disease. Cell, 148(6):1123–1131, 2012
2012
-
[72]
Alexander Wolf, Philipp Angerer, and Fabian J
F. Alexander Wolf, Philipp Angerer, and Fabian J. Theis. SCANPY: Large-scale single-cell gene expression data analysis.Genome Biology, 19(1):15, February 2018
2018
-
[73]
Random Matrix Theory-guided sparse PCA for single-cell RNA-seq data, September 2025
Victor Chardès. Random Matrix Theory-guided sparse PCA for single-cell RNA-seq data, September 2025
2025
-
[74]
An atlas of combinato- rial transcriptional regulation in mouse and man.Cell, 140(5):744–752, 2010
Timothy Ravasi, Harukazu Suzuki, Carlo Vittorio Can- nistraci, Shintaro Katayama, Vladimir B Bajic, Kai Tan, Altuna Akalin, Sebastian Schmeier, Mutsumi Kanamori- Katayama, Nicolas Bertin, et al. An atlas of combinato- rial transcriptional regulation in mouse and man.Cell, 140(5):744–752, 2010
2010
-
[75]
Belinda Phipson and Gordon K. Smyth. Permutation P- values Should Never Be Zero: Calculating Exact P-values When Permutations Are Randomly Drawn.Statistical Applications in Genetics and Molecular Biology, 9(1)
-
[76]
A connection between score matching and denoising autoencoders.Neural computation, 23(7):1661– 1674, 2011
Pascal Vincent. A connection between score matching and denoising autoencoders.Neural computation, 23(7):1661– 1674, 2011
2011
-
[77]
Generative modeling by estimating gradients of the data distribution.Advances in neural information processing systems, 32, 2019
Yang Song and Stefano Ermon. Generative modeling by estimating gradients of the data distribution.Advances in neural information processing systems, 32, 2019
2019
-
[78]
Bayesian learning via stochastic gradient langevin dynamics
Max Welling and Yee W Teh. Bayesian learning via stochastic gradient langevin dynamics. InProceedings of the 28th international conference on machine learning (ICML-11), pages 681–688, 2011. 15 Appendix A: Transport equation through conditional distributions In this section, we derive the continuity equation governing the time–evolving densitypt(x)induced...
2011
-
[79]
,xK−1), we recall the conditional Gaussian path considered by Lipman et al
Gaussian flow with constant and vanishing variance For a fixed conditioning variablezdefined with the tuple(x0, x1, . . . ,xK−1), we recall the conditional Gaussian path considered by Lipman et al. [43] (Theorem 3) as pt(x|z) =N x;µ t(z), σ 2 t (z) , t∈[0,1], where µt(z)and σt(z) > 0are differentiable in t. The mean conditional pathsµt are constructed in ...
-
[80]
Boundary conditions.This form also satisfies the appropriate boundary conditions given by the data at all times tk,∀k∈ [0,
Transport equation in the balanced setting Using the conditional distributions in the constant and vanishing variance limit derived in Corollary 1.1(a,b), we define the corresponding marginal distribution inxas pt(x) := Z pt(x|z)q(z)dz= Z δ(x−µ t(z))q(z)dz.(A2) 16 a. Boundary conditions.This form also satisfies the appropriate boundary conditions given by...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.