Functional and Density-Driven Errors in Density Functional Theory: Quantum Monte Carlo Benchmarks for Solids
Pith reviewed 2026-06-30 20:27 UTC · model grok-4.3
The pith
Quantum Monte Carlo reference densities show that functional errors dominate density-driven errors in most DFT calculations for solids but density-driven errors are larger by factors of two to three for some functionals.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
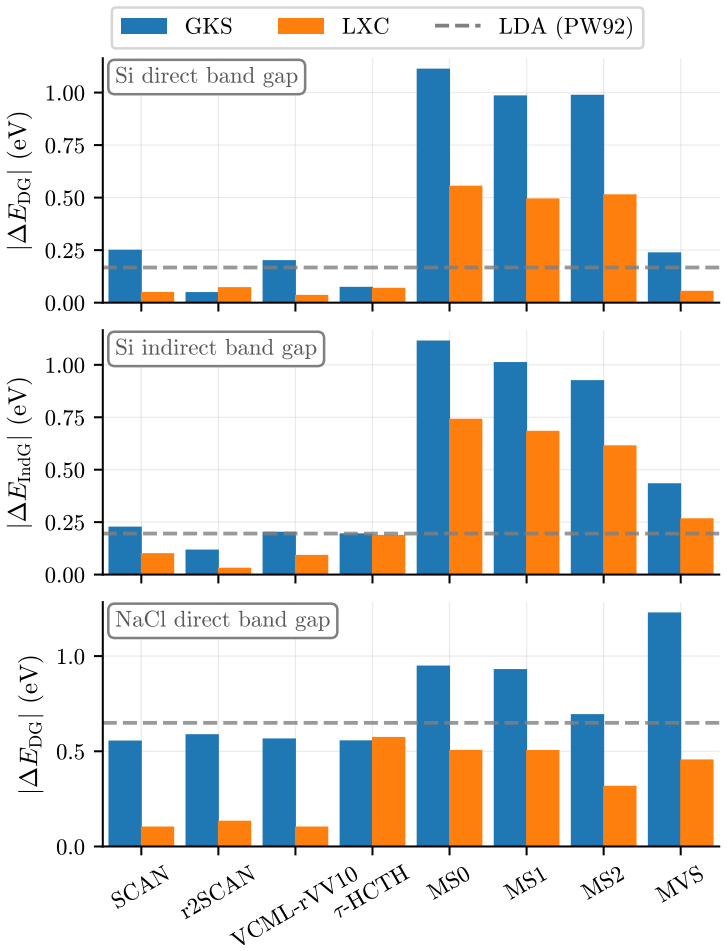
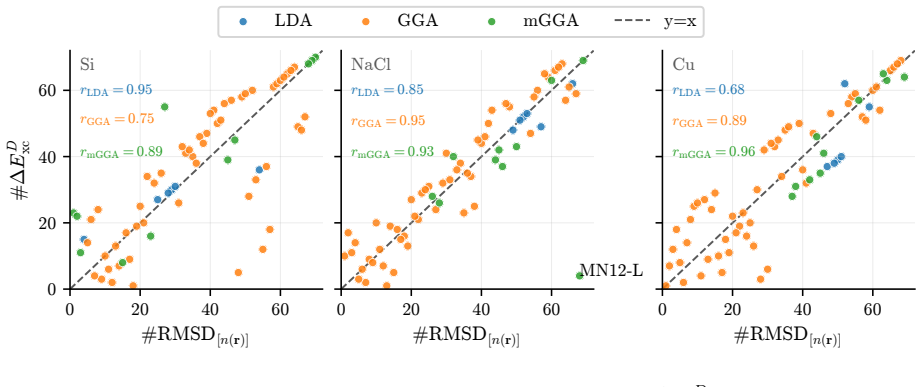
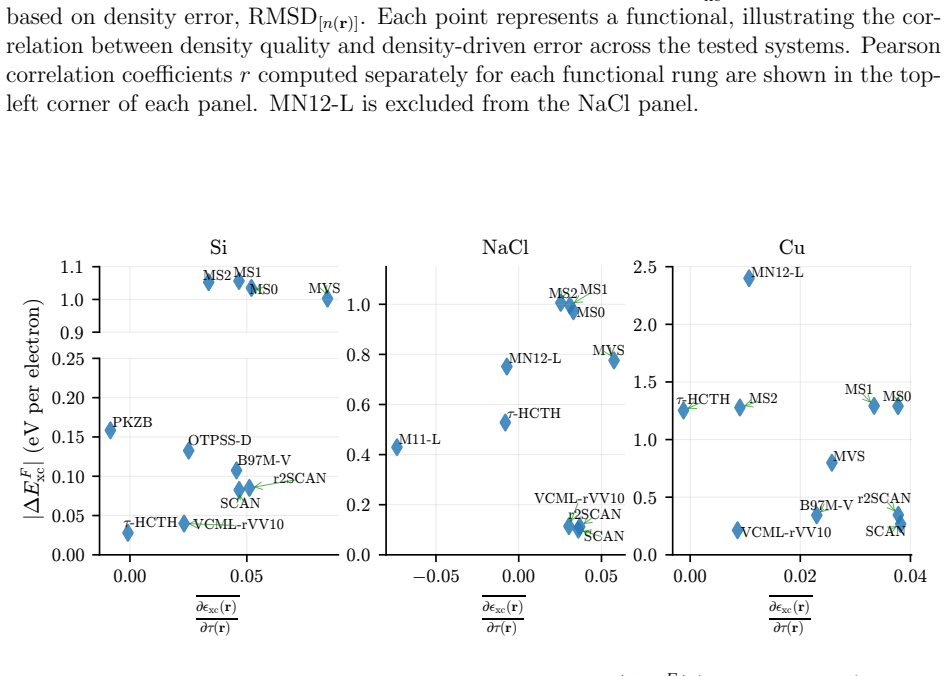
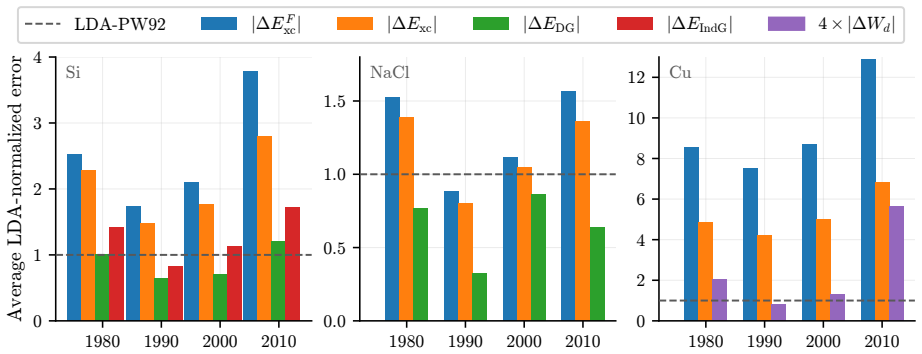
The central claim is that quantum Monte Carlo reference densities enable a quantitative split of DFT errors into functional and density components for three representative solids, demonstrating that functional errors predominate in most cases but density-driven errors can exceed them by factors of two to three for specific approximations in silicon and sodium chloride, while error cancellation is far more common in silicon than in copper and only a few functionals exceed LDA performance for copper even when supplied exact densities.
What carries the argument
The decomposition of total energy errors into functional-driven and density-driven parts by comparing self-consistent DFT calculations against non-self-consistent calculations on fixed quantum Monte Carlo densities.
If this is right
- Combinations of GILL or BECKE exchange with PBE, PW91, or P86 correlation yield near-exact exchange-correlation energies for silicon and sodium chloride when using QMC densities.
- Specialized functionals such as PBEsol or PBELYP are needed for copper to achieve good performance even with exact densities.
- High-quality densities reduce density-driven errors in all tested systems.
- Older 1990s GGA functionals often outperform modern meta-GGAs in these benchmarks.
- Material-dependent error cancellation may affect the transferability of machine-learned interatomic potentials.
Where Pith is reading between the lines
- Extending the analysis to additional solids could identify classes of materials where density-driven errors are systematically important.
- Developing density functionals specifically optimized for use with exact densities might improve accuracy beyond current approaches.
- These findings imply that error cancellation patterns should be checked when applying functionals across different material types in computational studies.
Load-bearing premise
The quantum Monte Carlo densities provide a sufficiently accurate reference to reliably distinguish and quantify functional-driven versus density-driven errors.
What would settle it
Recalculating the error separations using an independent high-accuracy density source, such as from coupled-cluster calculations or larger-scale quantum Monte Carlo, and finding that the relative magnitudes of density-driven and functional errors change for the identified exceptional functionals.
Figures
read the original abstract
We introduce a systematic analysis of density functional approximation errors in solids by separating functional-driven from density-driven contributions using quantum Monte Carlo densities of silicon, sodium chloride, and copper as reference. Typically, functional errors dominate, but we identify important exceptions where density-driven errors exceed functional errors by factors of 2-3, notably for SOGGA11 and {\tau}-HCTH in the semiconductor and the insulator. Material dependence is striking: 63% of functionals show error cancellation in silicon versus 18% in copper, and only five functionals surpass LDA accuracy for metallic copper even with exact densities. For silicon and sodium chloride, GILL or BECKE exchange combined with PBE, PW91, or P86 correlation achieves near-exact xc energies on QMC densities, while copper requires specialized functionals like PBEsol or PBELYP. High-quality densities consistently reduce density-driven errors across all systems. Historical analysis reveals that 1990s GGA functionals outperform many modern meta-GGAs, contradicting expectations of systematic improvement along Jacob's ladder. These results provide practical guidance for functional selection and highlight implications for machine learning potential development, where material-dependent error cancellation may compromise transferability.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a systematic decomposition of DFT approximation errors for solids into functional-driven and density-driven components, using QMC densities for Si, NaCl, and Cu as references. It reports that functional errors typically dominate but identifies exceptions (e.g., SOGGA11 and τ-HCTH) where density-driven errors exceed them by factors of 2-3; material dependence is emphasized via statistics on error cancellation (63% of functionals in Si vs. 18% in Cu) and performance rankings, with recommendations for specific functional combinations and notes on historical trends versus Jacob's ladder expectations.
Significance. If the QMC reference densities are validated as sufficiently accurate, the work provides concrete, material-specific guidance for functional selection in solids and highlights risks for transferability in machine-learned potentials arising from error cancellation patterns. The use of external QMC benchmarks for error separation is a methodological strength that enables falsifiable, quantitative claims without internal parameter fitting.
major comments (2)
- [Methods (QMC density generation and error decomposition procedure)] The quantitative separation of functional-driven (E_xc[n_QMC] minus true xc energy) versus density-driven (E_xc[n_self-consistent] minus E_xc[n_QMC]) errors, and all derived claims (2-3× dominance ratios, 63%/18% cancellation fractions, and functional rankings), rests on the unverified premise that QMC densities differ from true densities by amounts too small to contaminate the reported energy differences. No error bars on the QMC densities, no comparison to independent references (e.g., different nodal surfaces or GW densities), and no sensitivity analysis of how density perturbations propagate into the decomposed errors are provided, especially for metallic Cu where nodal errors are expected to be larger.
- [Abstract and Results (error decomposition)] Abstract and results sections: the error decomposition procedure is stated to yield clear numerical findings, yet no details are supplied on statistical uncertainties, convergence criteria, or data exclusion rules. This prevents independent verification that the central numbers (factors of 2-3, material-specific percentages) are robust rather than sensitive to analysis choices.
minor comments (1)
- [Introduction or Methods] Notation for the decomposed energy components should be defined explicitly with equations at first use to avoid ambiguity between total, functional-driven, and density-driven contributions.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review. The two major comments correctly identify gaps in the validation and reporting of the QMC reference data and numerical procedures. We address each point below and will incorporate the requested information in a revised manuscript.
read point-by-point responses
-
Referee: [Methods (QMC density generation and error decomposition procedure)] The quantitative separation of functional-driven (E_xc[n_QMC] minus true xc energy) versus density-driven (E_xc[n_self-consistent] minus E_xc[n_QMC]) errors, and all derived claims (2-3× dominance ratios, 63%/18% cancellation fractions, and functional rankings), rests on the unverified premise that QMC densities differ from true densities by amounts too small to contaminate the reported energy differences. No error bars on the QMC densities, no comparison to independent references (e.g., different nodal surfaces or GW densities), and no sensitivity analysis of how density perturbations propagate into the decomposed errors are provided, especially for metallic Cu where nodal errors are expected to be larger.
Authors: We agree that explicit validation of the QMC densities is essential for the reliability of the error decomposition. The original manuscript does not contain error bars on the densities, comparisons to alternative references, or a sensitivity analysis. In the revised version we will add a new subsection in Methods that (i) cites prior QMC benchmarks for the same Si, NaCl, and Cu systems against experiment and other high-level methods, (ii) reports the statistical uncertainties on the QMC densities obtained from the production runs, and (iii) presents a sensitivity test in which the input densities are perturbed by amounts consistent with those uncertainties; the resulting changes in the decomposed functional- and density-driven errors will be tabulated. For Cu we will explicitly discuss the fixed-node approximation and its expected influence on the metallic density. revision: yes
-
Referee: [Abstract and Results (error decomposition)] Abstract and results sections: the error decomposition procedure is stated to yield clear numerical findings, yet no details are supplied on statistical uncertainties, convergence criteria, or data exclusion rules. This prevents independent verification that the central numbers (factors of 2-3, material-specific percentages) are robust rather than sensitive to analysis choices.
Authors: We concur that the absence of these details limits reproducibility. The revised manuscript will expand the Methods section to specify (a) the statistical error bars on all QMC energies and densities, (b) the DFT convergence thresholds (energy, density, and k-point sampling) used for both self-consistent and non-self-consistent calculations, and (c) the precise criteria applied when averaging or selecting data points for the reported percentages and rankings. Error bars will be added to the relevant tables and figures, and a short paragraph will discuss how the quoted factors of 2–3 and the 63 % / 18 % cancellation statistics respond to reasonable variations in these choices. revision: yes
Circularity Check
Benchmark decomposition uses external QMC reference with no self-referential reductions
full rationale
The paper performs a computational benchmark separating functional-driven and density-driven DFT errors by evaluating exchange-correlation energies on QMC densities versus self-consistent densities for Si, NaCl, and Cu. No equations, parameters, or claims reduce by construction to fitted inputs or prior self-citations; the separation follows the standard external-reference protocol without renaming, ansatz smuggling, or load-bearing self-citation chains. The derivation chain is self-contained against the provided QMC data.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption QMC densities provide a reliable reference for decomposing DFT errors into functional-driven and density-driven components
Reference graph
Works this paper leans on
-
[1]
Inhomogeneous Electron Gas
(1) Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Physical Review1964, 136, B864–B871. (2) Kohn, W.; Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review1965, 140, A1133–A1138. (3) Prodan, E.; Kohn, W. Nearsightedness of electronic matter. Proceedings of the National Academy of Sciences2005, 102, 11635–11...
2018
-
[2]
F.; Levitt, A.; Canc` es, E
(29) Herbst, M. F.; Levitt, A.; Canc` es, E. DFTK: A Julian approach for simulating electrons in solids. Proc. JuliaCon Conf.2021, 3,
2021
-
[3]
(30) Tran, F.; Lehtola, S.; Pittalis, S.; Marques, M. A. L. Semi-Local Exchange-Correlation 31 Approximations in Density Functional Theory. 2026;https://arxiv.org/abs/2602. 17333. (31) Khanna, V.; Tribedi, S.; Kanungo, B.; Gavini, V.; Zimmerman, P. M. Bridges from Wavefunction Theory to Density Functional Theory. Annual Review of Physical Chemistry2026, 7...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.