ViroBench: Benchmarking Nucleotide Foundation Models on Viral Genomics Tasks
Pith reviewed 2026-06-29 22:28 UTC · model grok-4.3
The pith
Nucleotide foundation models degrade on phylogenetic and temporal shifts in viruses and decouple likelihood from function in generations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
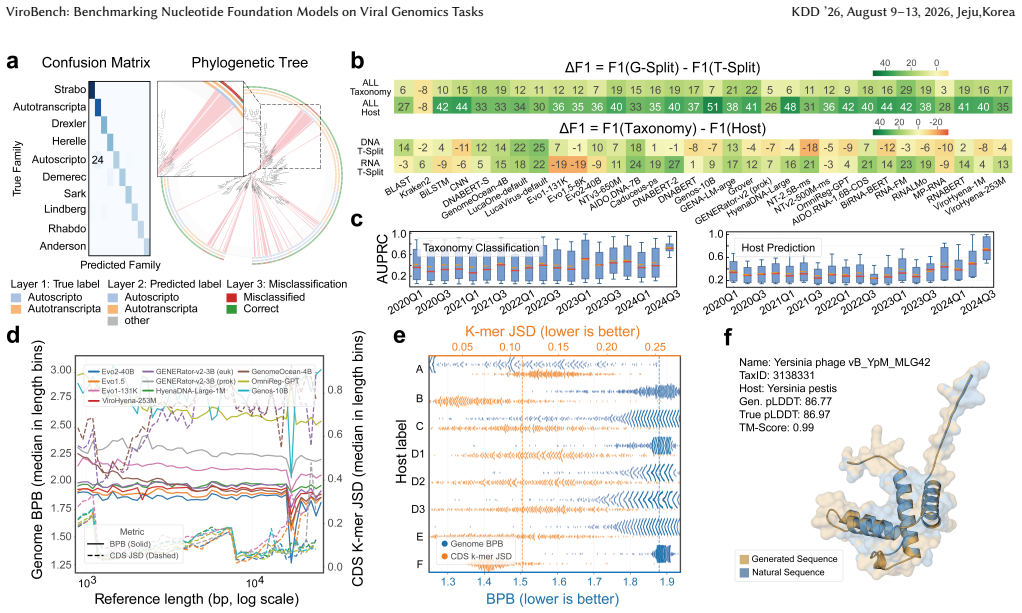
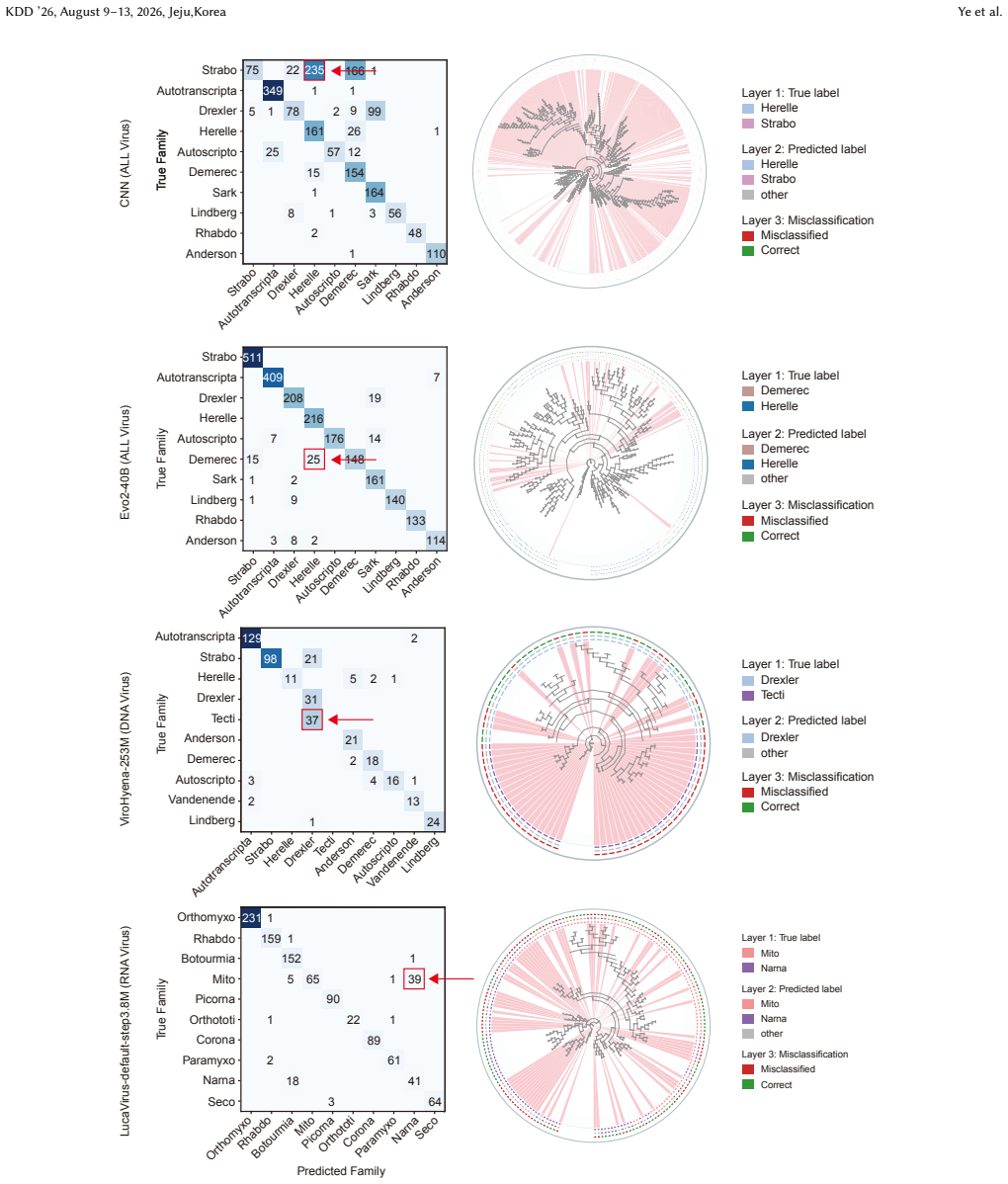
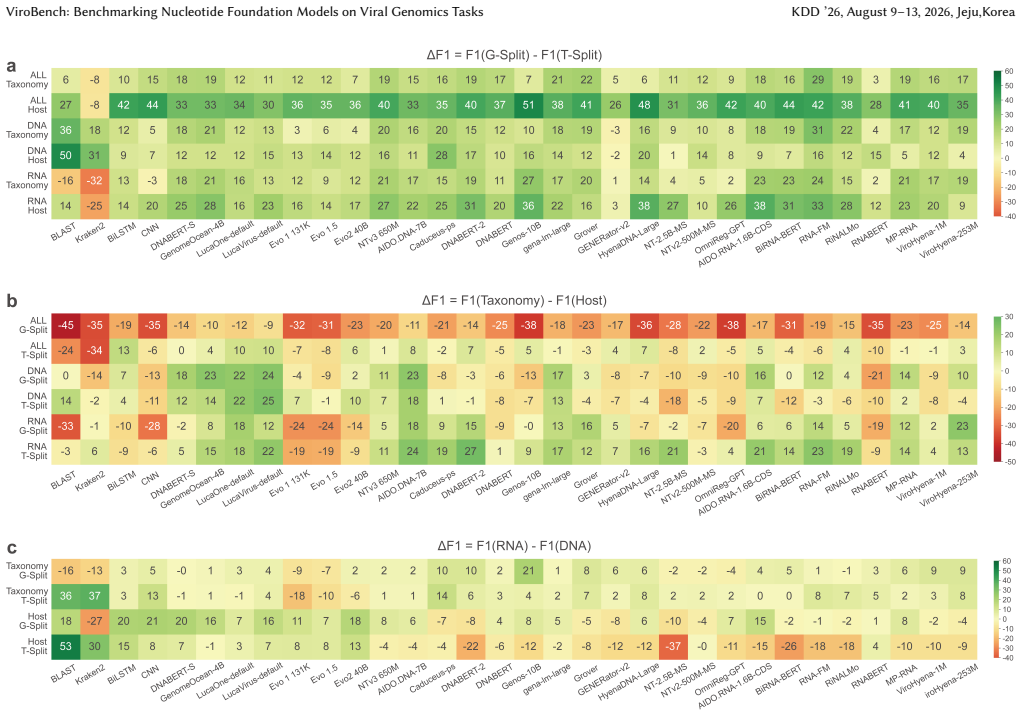
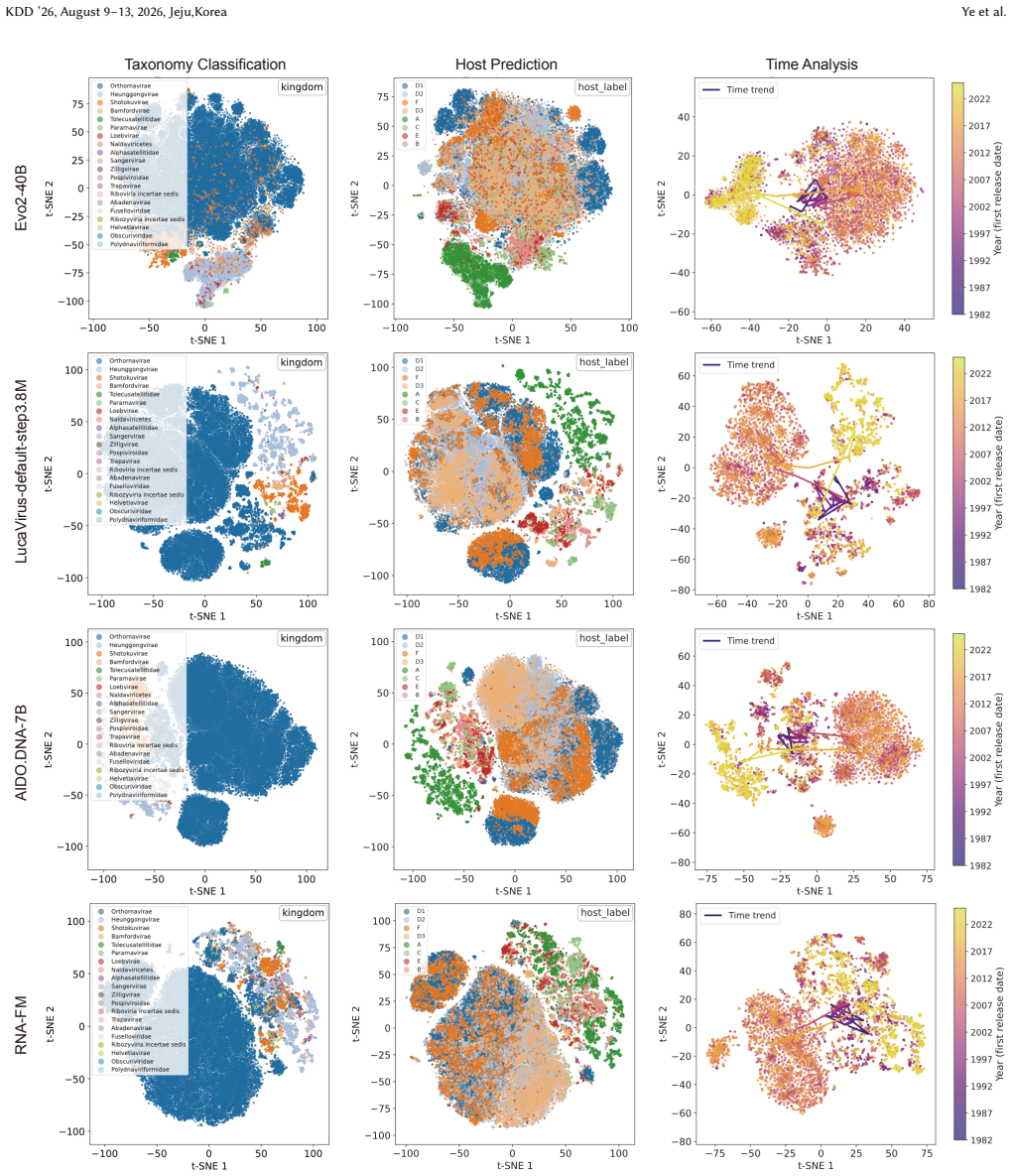
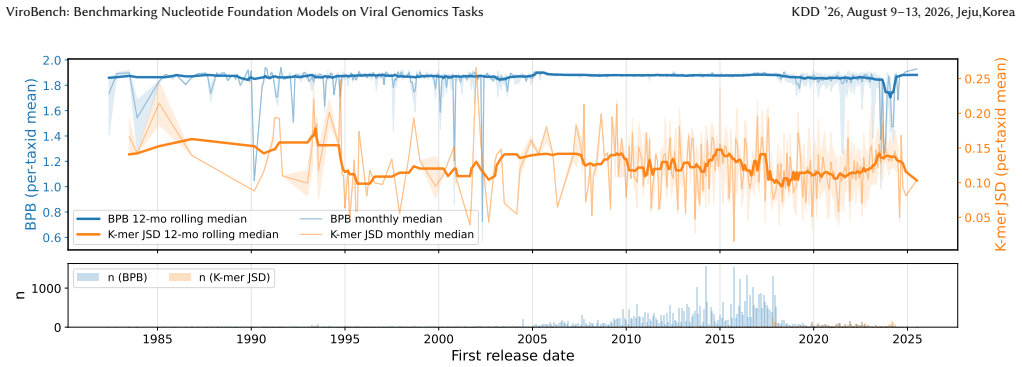
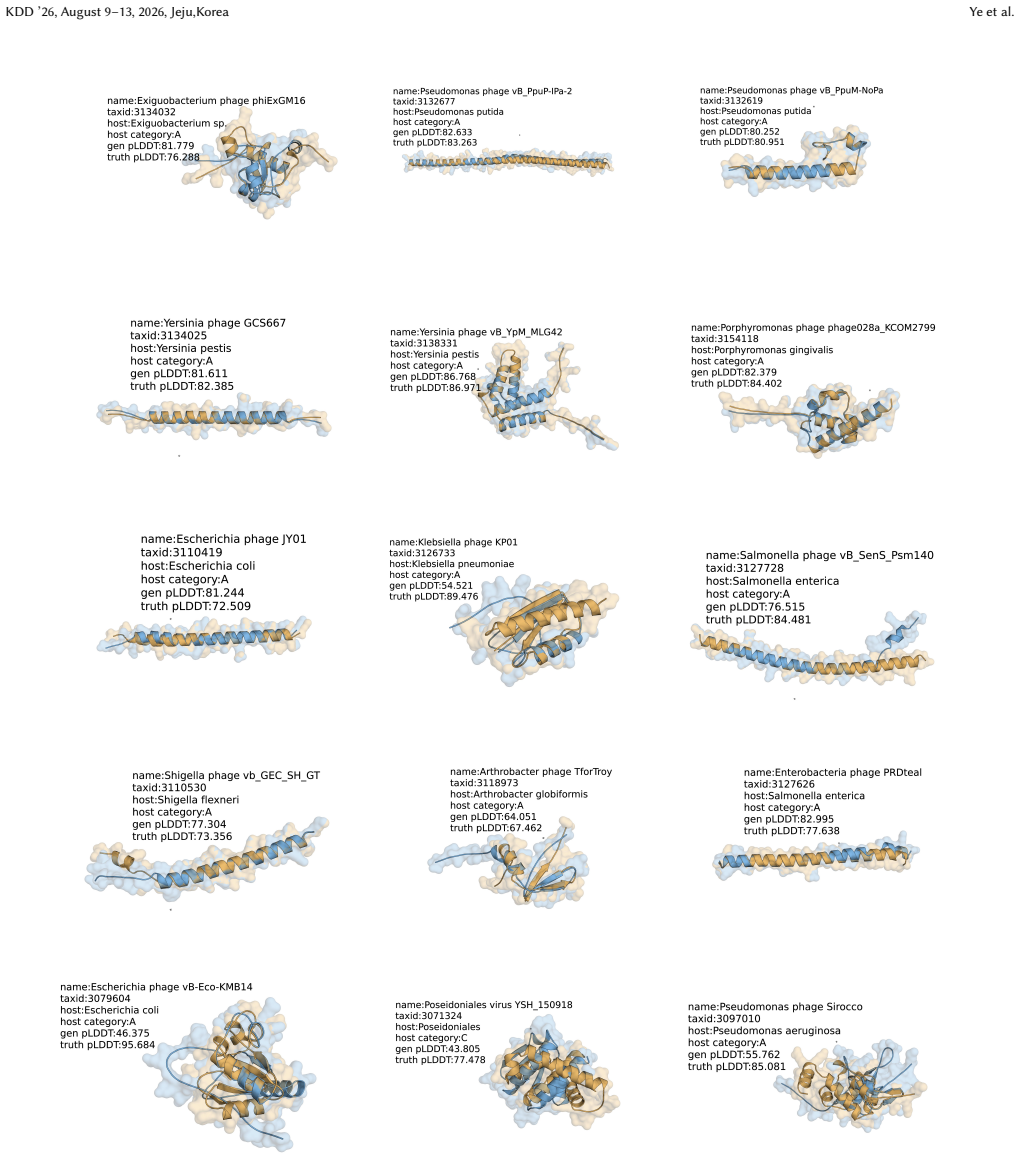
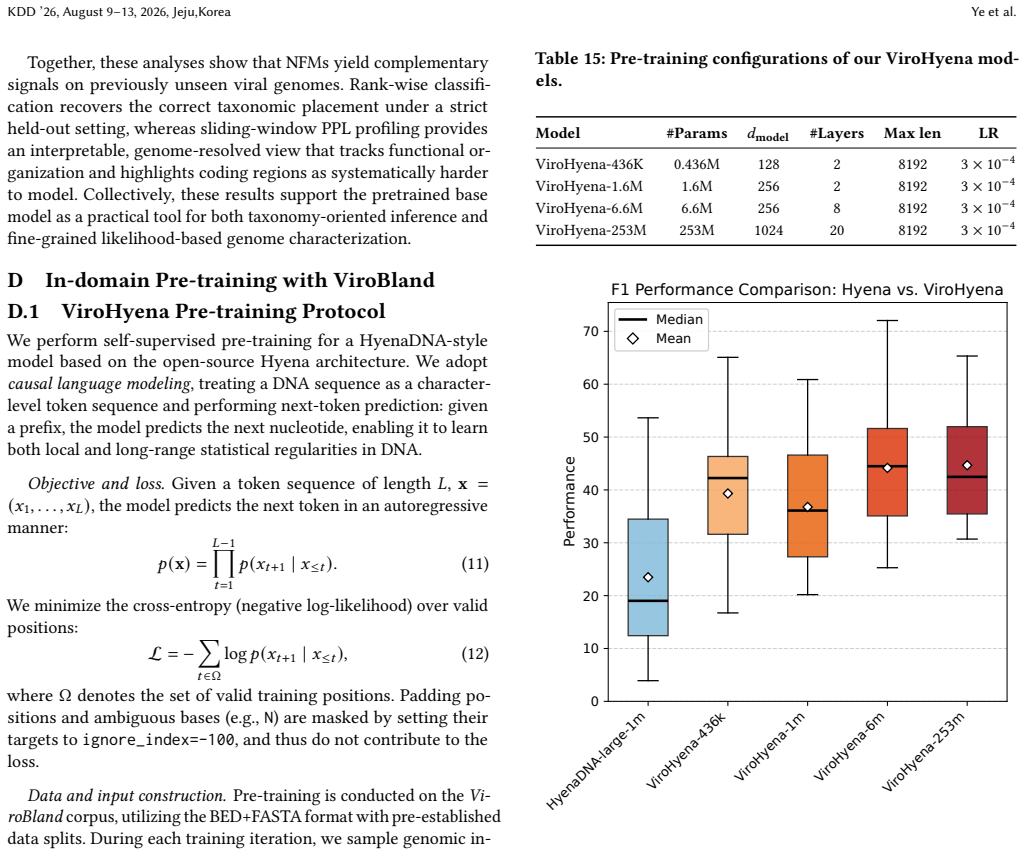
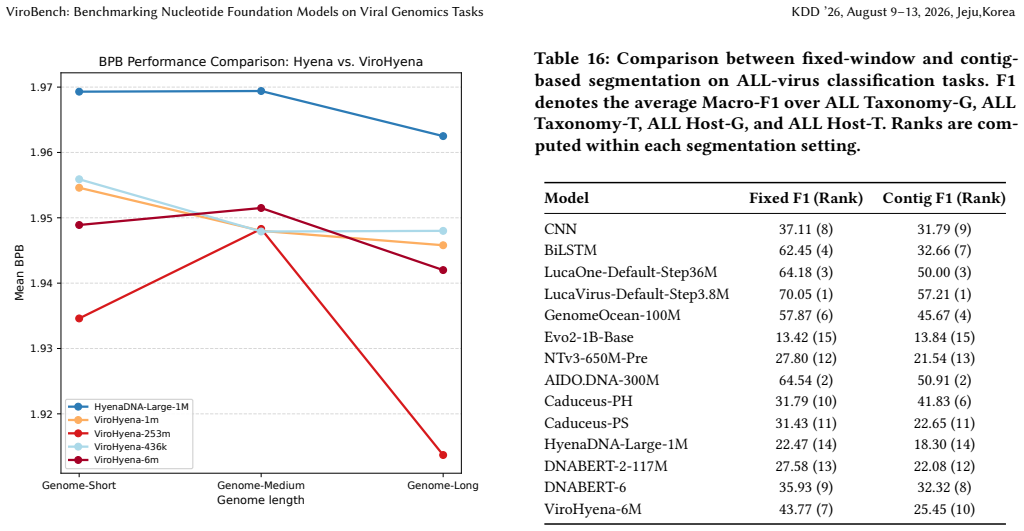
ViroBench demonstrates that NFMs exhibit a performance degradation in biological understanding under phylogenetic and temporal shifts, indicating weak extrapolation capabilities. Generation tasks reveal a decoupling between statistical likelihood and biological functional validity, posing latent biosecurity risks. Controlled ablation studies reveal that taxonomic diversity in pretraining data outweighs parameter scale, with a lightweight baseline achieving a 67.5% performance gain over its original model.
What carries the argument
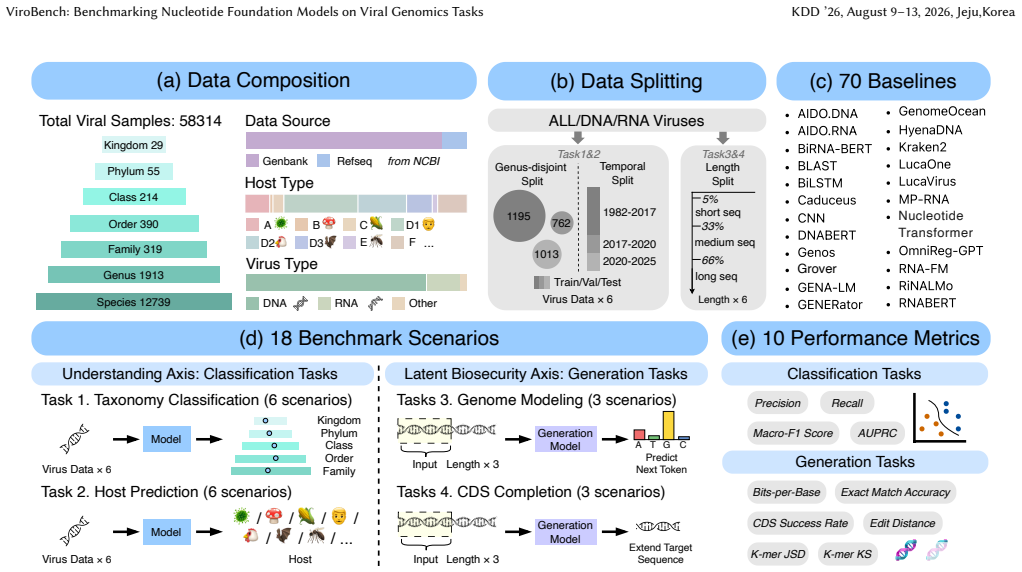
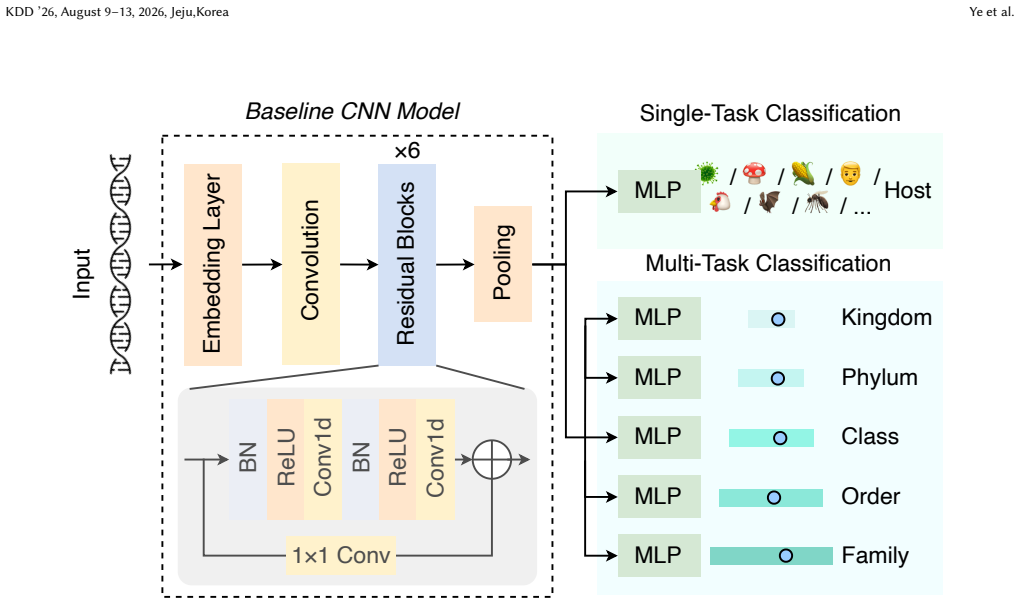
ViroBench benchmark with 18 scenarios across 4 task types assessing biological understanding and latent biosecurity risk in viral nucleotide models.
Load-bearing premise
The 18 scenarios and 4 task types in ViroBench sufficiently represent the critical dimensions of biological understanding and latent biosecurity risk for viral genomics applications.
What would settle it
Observing an NFM that maintains performance across phylogenetic and temporal shifts or where high-likelihood generations consistently match functional validity would falsify the degradation and decoupling claims.
Figures

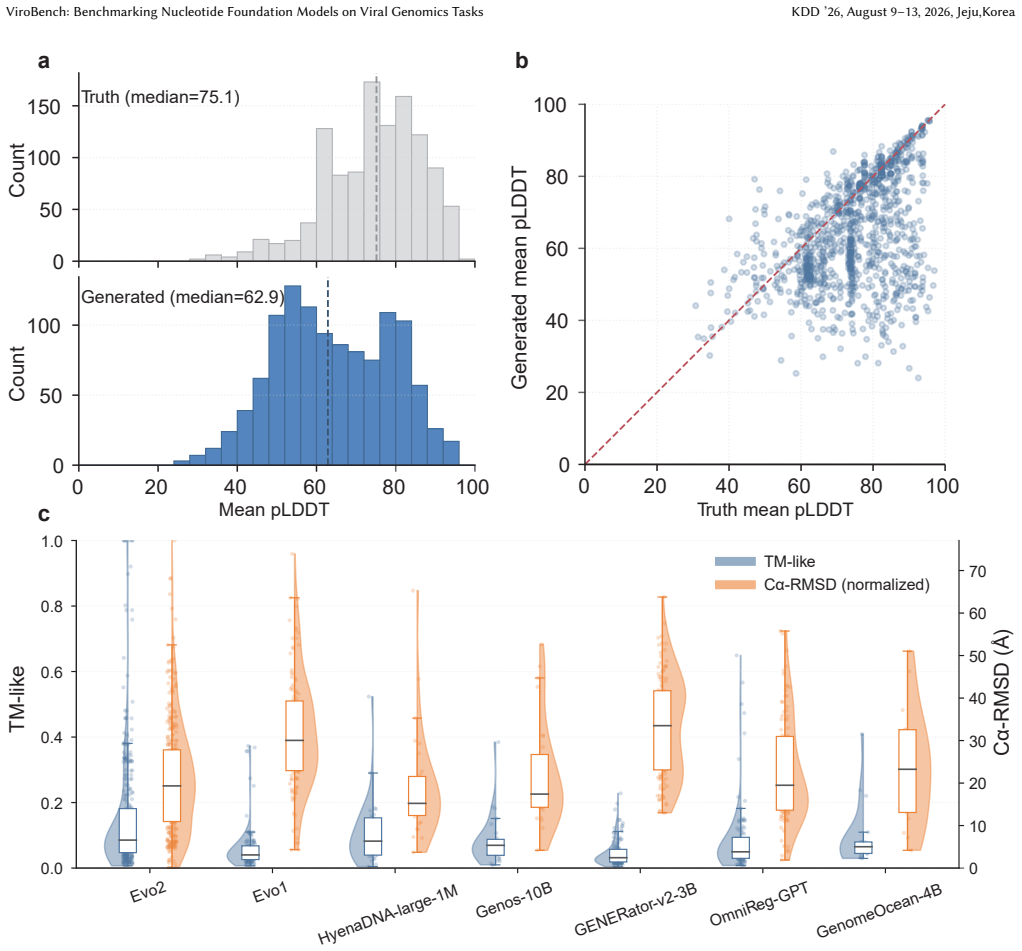
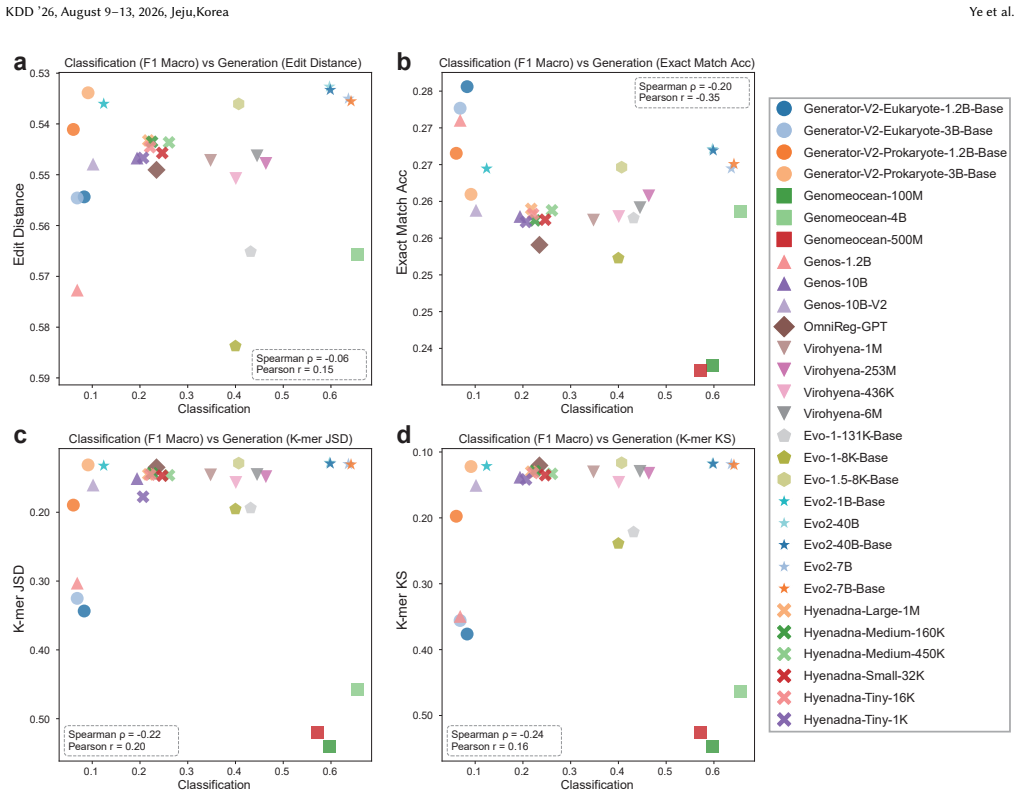
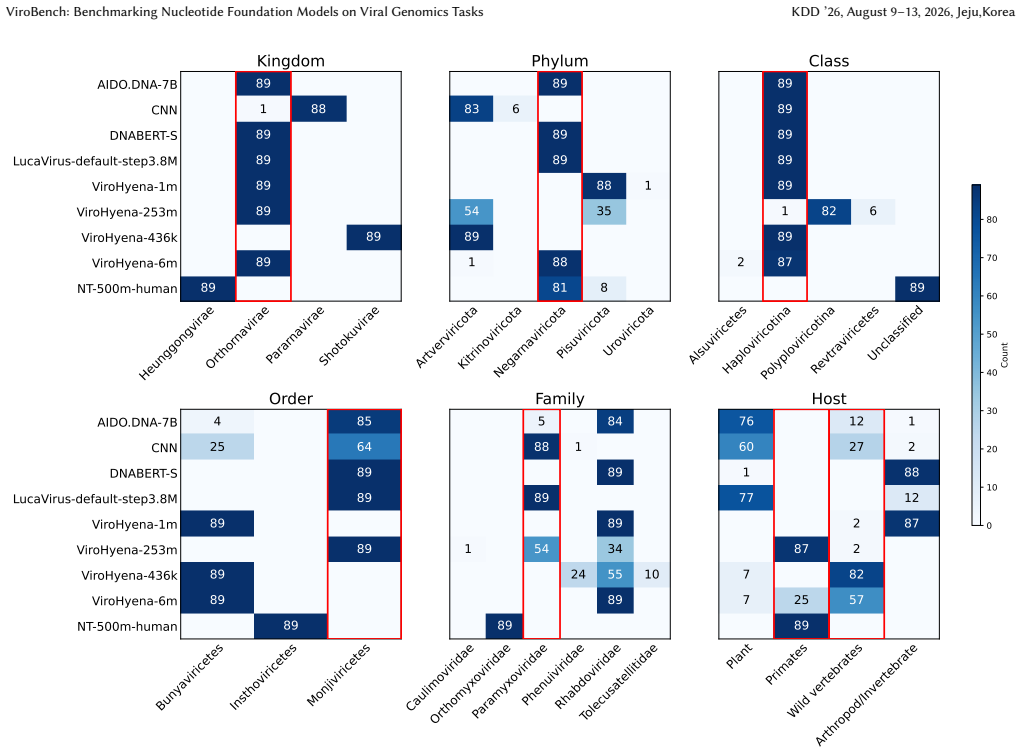
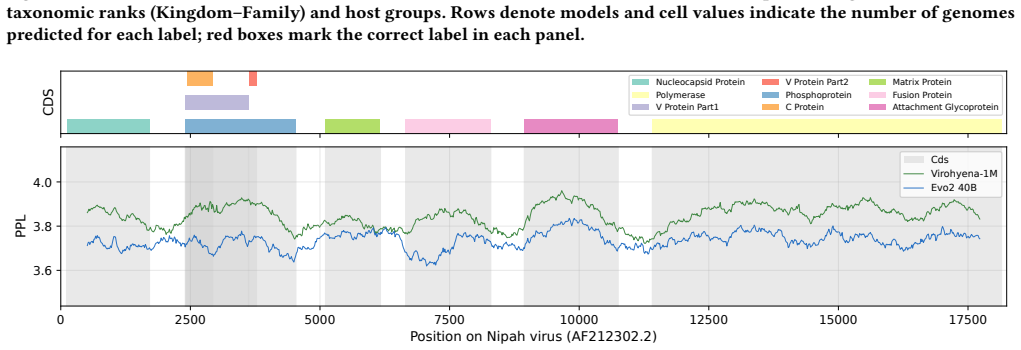
read the original abstract
Nucleotide sequences constitute the fundamental genetic basis of biological systems, rendering viral genomic analysis critical for biomedical advancement. Despite progress in biological foundation models, specifically nucleotide foundation models (NFMs), the field lacks a unified standard for viral genomics to facilitate community development and enforce biosecurity constraints. To address this, we introduce ViroBench, the first comprehensive and large-scale benchmark specifically designed for NFMs in viral settings. ViroBench evaluates models across two critical dimensions: biological understanding and latent biosecurity risk, covering 18 diverse scenarios within 4 task types. Extensive evaluation of 66 NFMs across diverse architectures yields three critical conclusions. Firstly, NFMs exhibit a performance degradation in biological understanding under phylogenetic and temporal shifts, indicating weak extrapolation capabilities. Secondly, generation tasks reveal a decoupling between statistical likelihood and biological functional validity, posing latent biosecurity risks. Thirdly, controlled ablation studies reveal that taxonomic diversity in pretraining data outweighs parameter scale. Specifically, a lightweight baseline trained on diverse data achieves a 67.5% performance gain over its original model. Overall, ViroBench provides interpretable, diagnostic evaluations and a reproducible measurement framework for future research on viral nucleotide foundation models. The datasets and code are publicly available at https://github.com/QIANJINYDX/ViroBench.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces ViroBench, the first large-scale benchmark for nucleotide foundation models (NFMs) on viral genomics. It evaluates 66 NFMs across 18 scenarios in 4 task types spanning biological understanding and latent biosecurity risk. The central claims are that NFMs show performance degradation under phylogenetic and temporal shifts, that generation tasks exhibit decoupling between statistical likelihood and functional validity (raising biosecurity concerns), and that taxonomic diversity in pretraining data outweighs parameter scale, evidenced by a lightweight baseline achieving a 67.5% performance gain. Datasets and code are released publicly.
Significance. If the benchmark tasks and evaluation protocols are sound, ViroBench would fill a clear gap by providing a reproducible, diagnostic framework for NFMs in viral settings. The public release of datasets and code is a concrete strength that supports community follow-up. The three conclusions, if substantiated, would usefully highlight generalization limits and safety considerations for future NFM development.
major comments (2)
- [Abstract] Abstract: the three conclusions are presented as resulting from 'extensive evaluation' of 66 models, yet the text supplies no description of the 18 scenarios, the four task types, the metrics, statistical tests, or error analysis. This absence is load-bearing because the degradation, decoupling, and diversity-over-scale claims cannot be assessed without those details.
- [Abstract] Abstract: the 67.5% performance gain for the lightweight baseline is stated without identifying the original model, the exact metric, the ablation controls, or the composition of the 'diverse data,' preventing evaluation of whether taxonomic diversity truly outweighs scale.
Simulated Author's Rebuttal
We thank the referee for their constructive feedback and for highlighting areas where the abstract could better support the manuscript's claims. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the three conclusions are presented as resulting from 'extensive evaluation' of 66 models, yet the text supplies no description of the 18 scenarios, the four task types, the metrics, statistical tests, or error analysis. This absence is load-bearing because the degradation, decoupling, and diversity-over-scale claims cannot be assessed without those details.
Authors: We agree the abstract is concise and omits these specifics. The main text details the 18 scenarios and four task types in Section 3, the metrics and evaluation protocols (including statistical tests) in Section 4, and error analysis in Section 5. To improve accessibility, we will revise the abstract to briefly name the task types and note the use of standard metrics with significance testing. revision: yes
-
Referee: [Abstract] Abstract: the 67.5% performance gain for the lightweight baseline is stated without identifying the original model, the exact metric, the ablation controls, or the composition of the 'diverse data,' preventing evaluation of whether taxonomic diversity truly outweighs scale.
Authors: The ablation details—including the original model, exact metric, controls, and diverse data composition—are reported in Section 5.3. We acknowledge the abstract lacks this context and will revise it to specify the model, metric, and key elements of the taxonomic diversity. revision: yes
Circularity Check
Empirical benchmark with no circular derivations
full rationale
This is a pure empirical benchmark paper that introduces ViroBench, evaluates 66 NFMs on 18 scenarios across 4 task types, and reports direct performance measurements. All three conclusions (degradation under shifts, likelihood-functional decoupling, and taxonomic diversity outweighing scale) are presented as observations from those measurements and ablations, with no equations, fitted parameters renamed as predictions, self-definitional steps, or load-bearing self-citations. The derivation chain is empty; results are self-contained as reported benchmark outcomes.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J Ballard, Joshua Bambrick, et al. 2024. Accurate structure prediction of biomolecular interactions with AlphaFold 3.Nature630, 8016 (2024), 493–500
2024
-
[2]
Manato Akiyama and Yasubumi Sakakibara. 2022. Informative RNA base em- bedding for RNA structural alignment and clustering by deep representation learning.NAR genomics and bioinformatics4, 1 (2022), lqac012
2022
-
[3]
P Balakrishnan, A Anny Leema, V Dhivya Shree, C Mohammad Saad, and A Mohan Babu. 2025. Gene-LLMs: a comprehensive survey of transformer-based genomic language models for regulatory and clinical genomics.Frontiers in Genetics16 (2025), 1634882
2025
-
[4]
Dennis A Benson, Mark Cavanaugh, Karen Clark, Ilene Karsch-Mizrachi, David J Lipman, James Ostell, and Eric W Sayers. 2012. GenBank.Nucleic acids research 41, D1 (2012), D36–D42
2012
-
[5]
Sam Boshar, Benjamin Evans, Ziqi Tang, Armand Picard, Yanis Adel, Franziska K Lorbeer, Chandana Rajesh, Tristan Karch, Shawn Sidbon, David Emms, et al
-
[6]
A foundational model for joint sequence-function multi-species modeling at scale for long-range genomic prediction.bioRxiv(2025), 2025–12
2025
-
[7]
Sebastian Bowyer, David J Allen, and Nicholas Furnham. 2025. Unveiling the ghost: machine learning’s impact on the landscape of virology.Journal of General Virology106, 1 (2025), 002067
2025
-
[8]
Garyk Brixi, Matthew G Durrant, Jerome Ku, Mohsen Naghipourfar, Michael Poli, Gwanggyu Sun, Greg Brockman, Daniel Chang, Alison Fanton, Gabriel A Gonzalez, et al. 2026. Genome modelling and design across all domains of life with Evo 2.Nature652, 8112 (2026), 1349–1361
2026
-
[9]
Christiam Camacho, George Coulouris, Vahram Avagyan, Ning Ma, Jason Pa- padopoulos, Kevin Bealer, and Thomas L Madden. 2009. BLAST+: architecture and applications.BMC bioinformatics10, 1 (2009), 421
2009
-
[10]
Jiayang Chen, Zhihang Hu, Siqi Sun, Qingxiong Tan, {WANG Yixuan}, Qinze Yu, Licheng Zong, Liang Hong, Jin Xiao, Irwin King, and {LI Yu}. 2022. Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions. The 2022 ICML Workshop on Computational Biology ; Conference date: 17-07-2022 Through 23-07-2022
2022
-
[11]
Hugo Dalla-Torre, Liam Gonzalez, Javier Mendoza-Revilla, Nicolas Lopez Car- ranza, Adam Henryk Grzywaczewski, Francesco Oteri, Christian Dallago, Evan Trop, Bernardo P de Almeida, Hassan Sirelkhatim, et al. 2025. Nucleotide Trans- former: building and evaluating robust foundation models for human genomics. Nature Methods22, 2 (2025), 287–297
2025
-
[12]
Christian Dallago, Jody Mou, Kadina E Johnston, Bruce Wittmann, Nick Bhat- tacharya, Samuel Goldman, Ali Madani, and Kevin K Yang. 2021. FLIP: Benchmark tasks in fitness landscape inference for proteins. InThirty-fifth Conference on Neural Information Processing Systems Datasets and Benchmarks Track (Round 2). https://openreview.net/forum?id=p2dMLEwL8tF
2021
-
[13]
Xing, and Le Song
Caleb Ellington, Ning Sun, Nicholas Ho, Tianhua Tao, Sazan Mahbub, Dian Li, Yonghao Zhuang, Hongyi Wang, Eric P. Xing, and Le Song. 2024. Accurate and General DNA Representations Emerge from Genome Foundation Models at Scale. InNeurIPS 2024 Workshop on AI for New Drug Modalities. https://openreview. net/forum?id=Kis8tVUeNi
2024
-
[14]
Alfred Ferrer Florensa, Jose Juan Almagro Armenteros, Henrik Nielsen, Frank Møller Aarestrup, and Philip Thomas Lanken Conradsen Clausen. 2024. SpanSeq: similarity-based sequence data splitting method for improved develop- ment and assessment of deep learning projects.NAR Genomics and Bioinformatics 6, 3 (2024), lqae106
2024
-
[15]
Veniamin Fishman, Yuri Kuratov, Aleksei Shmelev, Maxim Petrov, Dmitry Penzar, Denis Shepelin, Nikolay Chekanov, Olga Kardymon, and Mikhail Burtsev. 2025. GENA-LM: a family of open-source foundational DNA language models for long sequences.Nucleic Acids Research53, 2 (2025), gkae1310
2025
-
[16]
Zhangyang Gao, Cheng Tan, Yijie Zhang, Xingran Chen, Lirong Wu, and Stan Z Li. 2023. Proteininvbench: Benchmarking protein inverse folding on diverse tasks, models, and metrics.Advances in Neural Information Processing Systems36 (2023), 68207–68220
2023
-
[17]
Cody Glickman, Jo Hendrix, and Michael Strong. 2021. Simulation study and comparative evaluation of viral contiguous sequence identification tools.BMC bioinformatics22, 1 (2021), 329
2021
-
[18]
Katarína Grešová, Vlastimil Martinek, David Čechák, Petr Šimeček, and Panagi- otis Alexiou. 2023. Genomic benchmarks: a collection of datasets for genomic sequence classification.BMC Genomic Data24, 1 (2023), 25
2023
-
[19]
Yong He, Pan Fang, Yongtao Shan, Yuanfei Pan, Yanhong Wei, Yichang Chen, Yihao Chen, Yi Liu, Zhenyu Zeng, Zhan Zhou, et al. 2025. Generalized biological foundation model with unified nucleic acid and protein language.Nature Machine Intelligence(2025), 1–12
2025
-
[20]
Edward C Holmes. 2013. What can we predict about viral evolution and emer- gence?Current opinion in virology3, 2 (2013), 180–184
2013
-
[21]
Yanrong Ji, Zhihan Zhou, Han Liu, and Ramana V Davuluri. 2021. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome.Bioinformatics37, 15 (2021), 2112–2120
2021
- [22]
-
[23]
Adi Lin, Bin Xie, Cheng Ye, Cheng Wang, Duoyuan Chen, Ercheng Wang, Fanfeng Lu, Guirong Xue, Haiqiang Zhang, Jiajie Zhan, et al. 2025. Genos: a human-centric genomic foundation model.GigaScience14 (2025), giaf132
2025
-
[24]
LeAnn M Lindsey, Nicole L Pershing, Anisa Habib, Keith Dufault-Thompson, W Zac Stephens, Anne J Blaschke, Xiaofang Jiang, and Hari Sundar. 2025. The impact of tokenizer selection in genomic language models.Bioinformatics41, 9 (2025), btaf456
2025
- [25]
-
[26]
Frederikke Isa Marin, Felix Teufel, Marc Horlacher, Dennis Madsen, Dennis Pultz, Ole Winther, and Wouter Boomsma. 2024. BEND: Benchmarking DNA Language Models on Biologically Meaningful Tasks. InThe Twelfth International Conference on Learning Representations. https://openreview.net/forum?id=uKB4cFNQFg
2024
-
[27]
Aditi T Merchant, Samuel H King, Eric Nguyen, and Brian L Hie. 2026. Semantic design of functional de novo genes from a genomic language model.Nature649, 8097 (2026), 749–758
2026
-
[28]
Hayden C Metsky, Nicole L Welch, Priya P Pillai, Nicholas J Haradhvala, Laurie Rumker, Sreekar Mantena, Yibin B Zhang, David K Yang, Cheri M Ackerman, Juliane Weller, et al. 2022. Designing sensitive viral diagnostics with machine learning.Nature biotechnology40, 7 (2022), 1123–1131
2022
-
[29]
Florian Mock, Adrian Viehweger, Emanuel Barth, and Manja Marz. 2021. VIDHOP, viral host prediction with deep learning.Bioinformatics37, 3 (2021), 318–325
2021
-
[30]
Eric Nguyen, Michael Poli, Matthew G Durrant, Brian Kang, Dhruva Katrekar, David B Li, Liam J Bartie, Armin W Thomas, Samuel H King, Garyk Brixi, et al
-
[31]
Science386, 6723 (2024), eado9336
Sequence modeling and design from molecular to genome scale with Evo. Science386, 6723 (2024), eado9336
2024
-
[32]
Eric Nguyen, Michael Poli, Marjan Faizi, Armin Thomas, Michael Wornow, Cal- lum Birch-Sykes, Stefano Massaroli, Aman Patel, Clayton Rabideau, Yoshua Bengio, et al. 2023. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution.Advances in neural information processing systems36 (2023), 43177–43201
2023
-
[33]
Pascal Notin, Aaron Kollasch, Daniel Ritter, Lood Van Niekerk, Steffanie Paul, Han Spinner, Nathan Rollins, Ada Shaw, Rose Orenbuch, Ruben Weitzman, et al
-
[34]
Proteingym: Large-scale benchmarks for protein fitness prediction and design.Advances in neural information processing systems36 (2023), 64331–64379
2023
-
[35]
Nuala A O’Leary, Mathew W Wright, J Rodney Brister, Stacy Ciufo, Diana Haddad, Rich McVeigh, Bhanu Rajput, Barbara Robbertse, Brian Smith-White, Danso Ako- Adjei, et al. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation.Nucleic acids research44, D1 (2016), D733–D745
2016
-
[36]
Yuan-Fei Pan, Yong He, Yu-Qi Liu, Yong-Tao Shan, Shu-Ning Liu, Jia-Hao Ma, Xue Liu, Xiaoyun Pan, Yinqi Bai, Zan Xu, et al. 2025. Predicting the evolutionary and functional landscapes of viruses with a unified nucleotide-protein language model: Lucavirus.bioRxiv(2025), 2025–06
2025
-
[37]
Rafael Josip Penić, Tin Vlašić, Roland G Huber, Yue Wan, and Mile Šikić. 2025. Rinalmo: General-purpose rna language models can generalize well on structure prediction tasks.Nature Communications16, 1 (2025), 5671
2025
-
[38]
Fedor S Perelygin, Alexander N Lukashev, and Yulia A Aleshina. 2025. The effect of taxonomic, host-dependent features and sample bias on virus host prediction using machine learning and short sequence k-mers.Scientific reports15, 1 (2025), 31592
2025
-
[39]
Purwono Purwono, Annastasya Nabila Elsa Wulandari, and Novieta Hardeani Sari. 2024. Virus Host Prediction with Metagenomic Features using Support Vec- tor Machine Algorithm and Grid Search Cross Validation Optimization.Journal of Advanced Health Informatics Research2, 3 (2024), 127–137
2024
-
[40]
Rajan Saha Raju, Abdullah Al Nahid, Preonath Chondrow Dev, and Rashedul Islam. 2022. VirusTaxo: Taxonomic classification of viruses from the genome sequence using k-mer enrichment.Genomics114, 4 (2022), 110414
2022
-
[41]
Jie Ren, Nathan A Ahlgren, Yang Young Lu, Jed A Fuhrman, and Fengzhu Sun
-
[42]
VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data.Microbiome5, 1 (2017), 69
2017
-
[43]
Jie Ren, Kai Song, Chao Deng, Nathan A Ahlgren, Jed A Fuhrman, Yi Li, Xiaohui Xie, Ryan Poplin, and Fengzhu Sun. 2020. Identifying viruses from metagenomic data using deep learning.Quantitative Biology8, 1 (2020), 64–77
2020
-
[44]
Melissa Sanabria, Jonas Hirsch, Pierre M Joubert, and Anna R Poetsch. 2024. DNA language model GROVER learns sequence context in the human genome.Nature Machine Intelligence6, 8 (2024), 911–923
2024
-
[45]
Rafael Sanjuán, Miguel R Nebot, Nicola Chirico, Louis M Mansky, and Robert Belshaw. 2010. Viral mutation rates.Journal of virology84, 19 (2010), 9733–9748
2010
-
[46]
Josep Sardanyés, Celia Perales, Esteban Domingo, and Santiago F Elena. 2024. Quasispecies theory and emerging viruses: challenges and applications.npj Viruses2, 1 (2024), 54. ViroBench: Benchmarking Nucleotide Foundation Models on Viral Genomics Tasks KDD ’26, August 9–13, 2026, Jeju,Korea
2024
-
[47]
Yair Schiff, Chia Hsiang Kao, Aaron Gokaslan, Tri Dao, Albert Gu, and Volodymyr Kuleshov. 2024. Caduceus: Bi-Directional Equivariant Long-Range DNA Sequence Modeling. InProceedings of the 41st International Conference on Machine Learning (Proceedings of Machine Learning Research, Vol. 235), Ruslan Salakhutdinov, Zico Kolter, Katherine Heller, Adrian Welle...
2024
-
[48]
Mike Schuster and Kuldip K Paliwal. 1997. Bidirectional recurrent neural net- works.IEEE transactions on Signal Processing45, 11 (1997), 2673–2681
1997
-
[49]
Jiayu Shang and Yanni Sun. 2021. CHEER: HierarCHical taxonomic classification for viral mEtagEnomic data via deep leaRning.Methods189 (2021), 95–103
2021
-
[50]
Md Toki Tahmid, Haz Sameen Shahgir, Sazan Mahbub, Yue Dong, and Md Sham- suzzoha Bayzid. 2025. BiRNA-BERT allows efficient RNA language modeling with adaptive tokenization.Communications Biology8, 1 (2025), 1621
2025
-
[51]
Aowen Wang, Jiaqi Li, Hongyu Dong, Bocheng Xu, Qingyu Yin, Yanchao Xu, Jie Fu, and Junbo Zhao. 2025. Omnireg-gpt: a high-efficiency foundation model for comprehensive genomic sequence understanding.Nature Communications16, 1 (2025), 10139
2025
-
[52]
James D Watson and Francis HC Crick. 1953. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid.Nature171, 4356 (1953), 737–738
1953
-
[53]
Nicole E Wheeler. 2025. Responsible AI in biotechnology: balancing discovery, innovation and biosecurity risks.Frontiers in Bioengineering and Biotechnology 13 (2025), 1537471
2025
-
[54]
Derrick E Wood, Jennifer Lu, and Ben Langmead. 2019. Improved metagenomic analysis with Kraken 2.Genome biology20, 1 (2019), 257
2019
- [55]
-
[56]
Minghao Xu, Zuobai Zhang, Jiarui Lu, Zhaocheng Zhu, Yangtian Zhang, Ma Chang, Runcheng Liu, and Jian Tang. 2022. Peer: a comprehensive and multi-task benchmark for protein sequence understanding.Advances in Neural Information Processing Systems35 (2022), 35156–35173
2022
-
[57]
An Yang, Anfeng Li, Baosong Yang, Beichen Zhang, Binyuan Hui, Bo Zheng, Bowen Yu, Chang Gao, Chengen Huang, Chenxu Lv, et al. 2025. Qwen3 technical report.arXiv preprint arXiv:2505.09388(2025)
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[58]
Heng Yang and Ke Li. 2024. MP-RNA: Unleashing Multi-species RNA Foun- dation Model via Calibrated Secondary Structure Prediction. InFindings of the Association for Computational Linguistics: EMNLP 2024. 5278–5296
2024
-
[59]
Francisco Murilo Zerbini, Stuart G Siddell, Elliot J Lefkowitz, Arcady R Mushegian, Evelien M Adriaenssens, Poliane Alfenas-Zerbini, Donald M Dempsey, Bas E Dutilh, Maria Laura Garcia, R Curtis Hendrickson, et al. 2023. Changes to virus taxonomy and the ICTV Statutes ratified by the International Committee on Taxonomy of Viruses (2023).Archives of virolog...
2023
-
[60]
Liang Zhang, Hua Pang, Chenghao Zhang, Song Li, Yang Tan, Fan Jiang, Mingchen Li, Yuanxi Yu, Ziyi Zhou, Banghao Wu, et al . 2025. VenusMutHub: a systematic evaluation of protein mutation effect predictors on small-scale experimental data.Acta Pharmaceutica Sinica B(2025)
2025
-
[61]
Zhihan Zhou, Yanrong Ji, Weijian Li, Pratik Dutta, Ramana Davuluri, and Han Liu
-
[62]
InInternational Conference on Learning Representations, Vol
DNABERT-2: Efficient foundation model and benchmark for multi-species genomes. InInternational Conference on Learning Representations, Vol. 2024. 41642–41665
2024
-
[63]
Zhihan Zhou, Robert Riley, Satria Kautsar, Weimin Wu, Rob Egan, Steven Hofmeyr, Shira Goldhaber-Gordon, Mutian Yu, Harrison Ho, Fengchen Liu, et al
-
[64]
GenomeOcean: An Efficient Genome Foundation Model Trained on Large- Scale Metagenomic Assemblies.bioRxiv(2025), 2025–01
2025
-
[65]
Zhihan Zhou, Weimin Wu, Harrison Ho, Jiayi Wang, Lizhen Shi, Ramana V Davuluri, Zhong Wang, and Han Liu. 2025. DNABERT-S: Pioneering species differentiation with species-aware DNA embeddings.Bioinformatics41, Supple- ment_1 (2025), i255–i264
2025
-
[66]
Shuxian Zou, Tianhua Tao, Sazan Mahbub, Caleb Ellington, Robin Jonathan Algayres, Dian Li, Yonghao Zhuang, Hongyi Wang, Le Song, and Eric P. Xing
-
[67]
recorded time
A Large-Scale Foundation Model for RNA Function and Structure Prediction. InNeurIPS 2024 Workshop on AI for New Drug Modalities. https://openreview. net/forum?id=Gzo3JMPY8w Appendix Table of Contents A Detailed Data Curation Pipeline 12 A.1 Data Sources and Quality Filters 12 A.2 Data Partitioning 12 A.3 Recommended Lightweight Evaluation Subset 13 B Impl...
2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.