M\=oLe-{Λ}: Learning the Coupled-Cluster Response State for Energies, Gradients, and Properties
Pith reviewed 2026-06-29 08:25 UTC · model grok-4.3
The pith
A neural network jointly predicts the T and Λ amplitudes of CCSD to obtain energies, forces, and response properties from Hartree-Fock orbitals.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
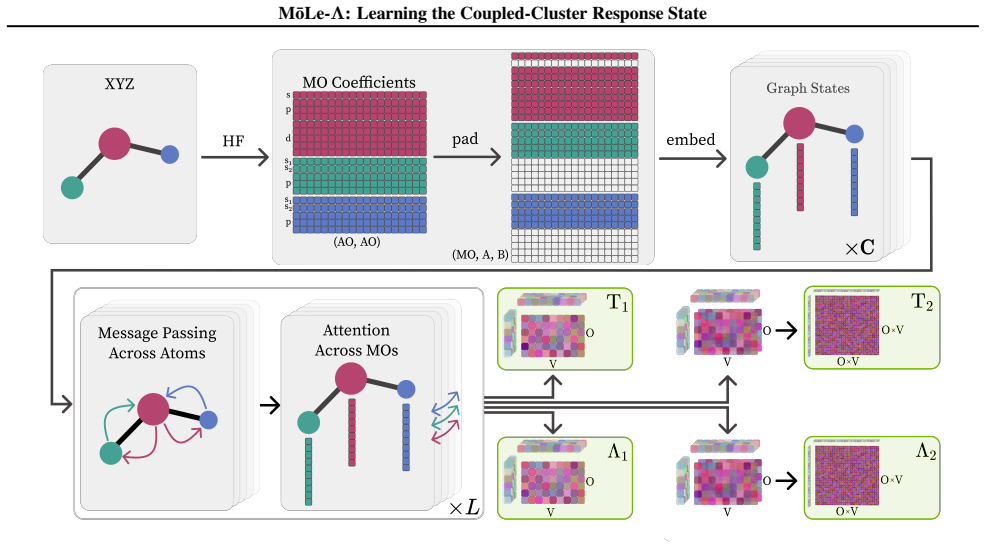
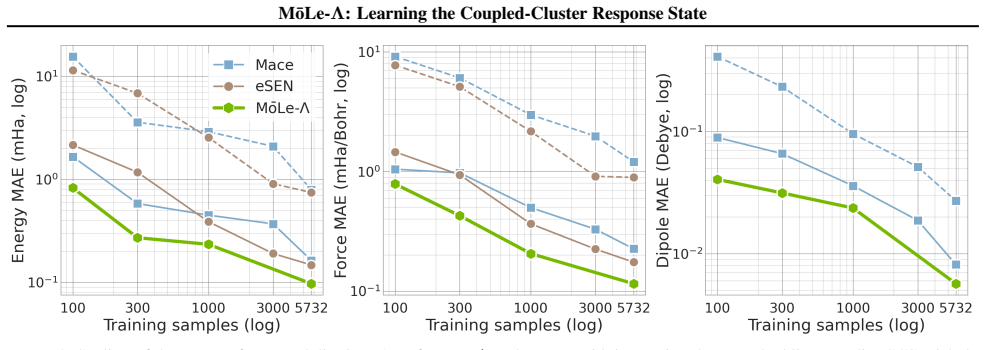
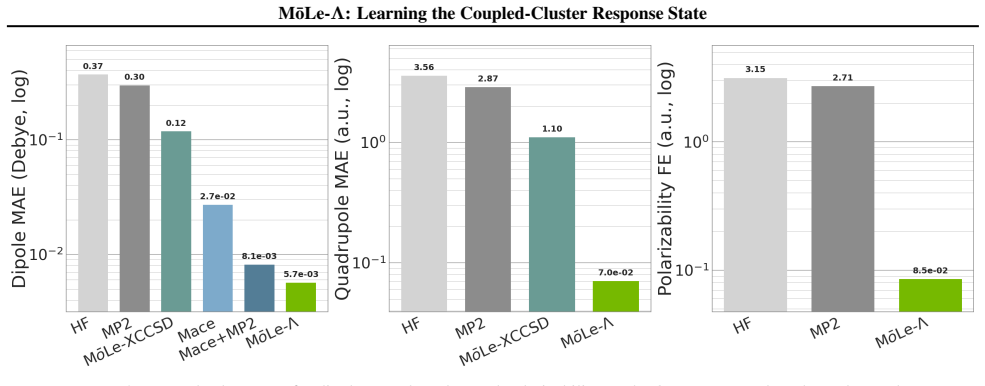
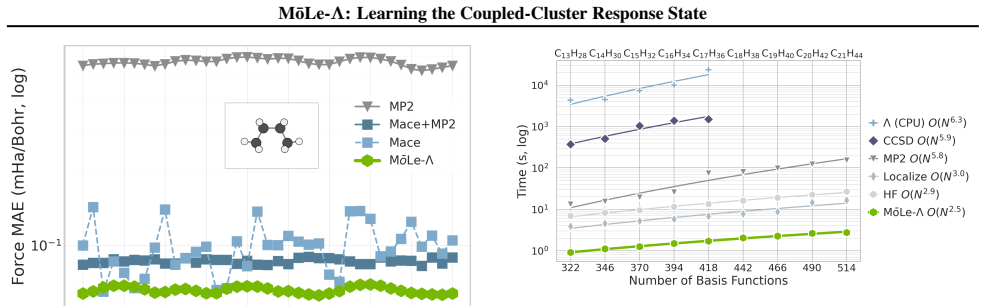
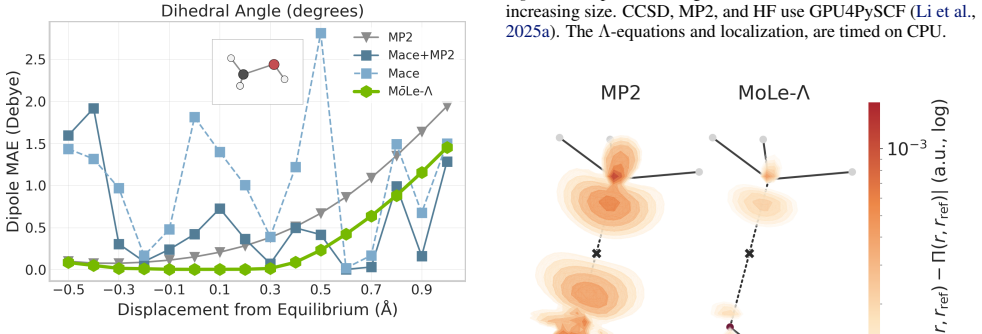
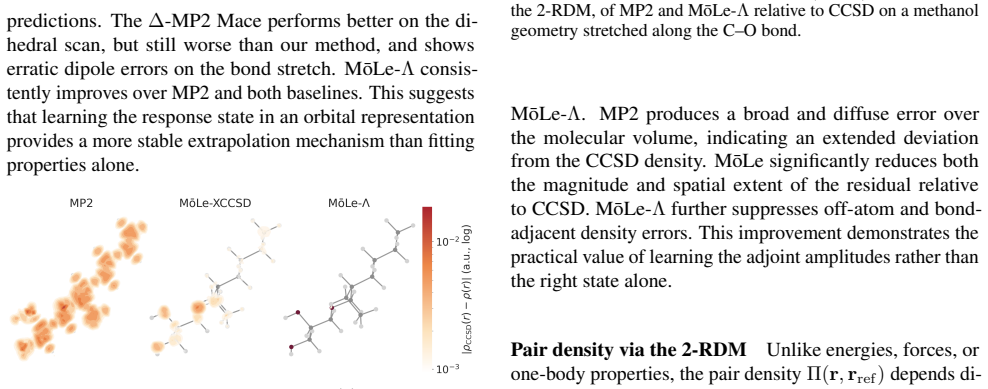
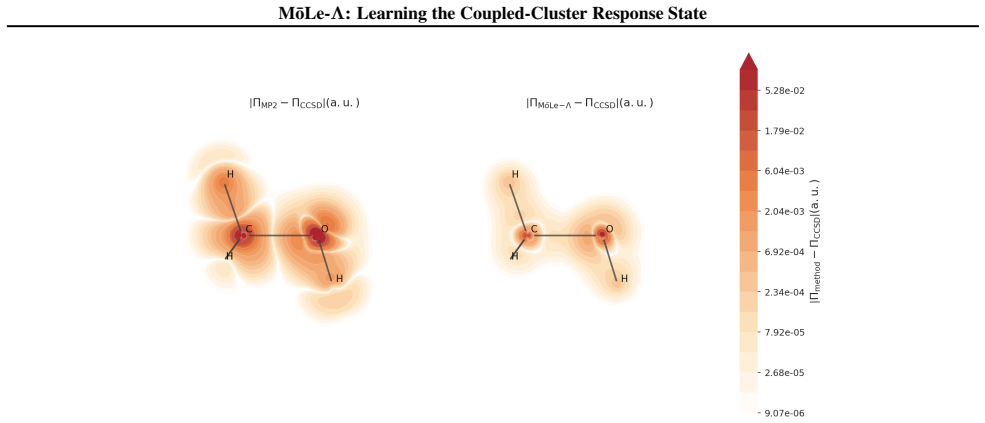
MōLe-Λ predicts the full ground-state coupled-cluster singles and doubles (CCSD) response state by jointly learning right-hand amplitudes (T1, T2) and left-hand amplitudes (Λ1, Λ2) from localized Hartree-Fock molecular orbitals. The model yields accurate CC-quality energies and forces while simultaneously recovering dipoles, quadrupoles, polarizabilities, the electron density, and 2-electron observables such as the pair density, extending the speed advantage of MōLe over full CCSD.
What carries the argument
The MōLe-Λ model with additional Λ1 and Λ2 readouts mirroring the symmetry constraints of the T1 and T2 heads, preserving the equivariant orbital encoder and size-extensivity.
If this is right
- Accurate CC-quality energies and forces are obtained alongside multiple response properties.
- The model recovers the electron density and pair density at CCSD level.
- It extends the speed advantage of the original MōLe over full CCSD while expanding accessible properties.
- Provides a route to wavefunction-level surrogate models for correlated quantum chemistry.
Where Pith is reading between the lines
- If the approach transfers across chemical space, it could support property calculations on molecules larger than those feasible with direct CCSD.
- The joint learning of T and Λ might enable consistent treatment of properties that depend on both amplitudes without separate models.
- Size-extensivity preserved by the architecture suggests applicability to extended systems.
Load-bearing premise
That training on localized Hartree-Fock molecular orbitals is sufficient to recover the full CCSD response state with transferability across chemical space without post-hoc corrections or molecule-specific adjustments.
What would settle it
A direct comparison showing that MōLe-Λ predicted polarizabilities or pair densities deviate substantially from CCSD reference values on molecules outside the training distribution.
Figures
read the original abstract
Coupled-cluster (CC) theory is often considered the gold standard of quantum chemistry, but its high computational cost limits routine access to accurate energies, forces and response properties. While the right-hand $T$-amplitudes determine the correlated wavefunction, many practically important observables additionally require the left-hand $\Lambda$-amplitudes. We introduce M\=oLe-$\Lambda$, an extension of Molecular Orbital Learning (M\=oLe) that predicts the full ground-state coupled-cluster singles and doubles (CCSD) response state by jointly learning right-hand amplitudes $(T_1,T_2)$ and left-hand amplitudes $(\Lambda_1,\Lambda_2)$ from localized Hartree--Fock molecular orbitals. Architecturally, M\=oLe-$\Lambda$ extends M\=oLe with $\Lambda_1$ and $\Lambda_2$ readouts that mirror the symmetry constraints of the $T_1$ and $T_2$ heads, while preserving the original equivariant orbital encoder, odd sign-equivariant decoding, locality and size-extensivity. The resulting model yields accurate CC-quality energies and forces, while simultaneously recovering dipoles, quadrupoles, polarizabilities, the electron density, and 2-electron observables such as the pair density. We show that M\=oLe-$\Lambda$ further extends the speed advantage of M\=oLe over full CCSD while substantially expanding the accessible properties, providing a route to wavefunction-level surrogate models for correlated quantum chemistry.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces MōLe-Λ as an extension of the MōLe framework that jointly learns the right-hand CCSD amplitudes (T1, T2) and left-hand amplitudes (Λ1, Λ2) from localized Hartree-Fock molecular orbitals. It claims that the resulting equivariant, size-extensive model delivers CC-quality energies and forces while also recovering dipoles, quadrupoles, polarizabilities, the electron density, and the pair density, thereby expanding the set of accessible properties beyond the original MōLe model.
Significance. If the numerical results and transferability claims hold, the work would be significant because it supplies a single learned surrogate for the full CCSD response state (both T and Λ), enabling efficient access to a broad suite of one- and two-electron properties that normally require separate left- and right-hand solutions. This would constitute a concrete step toward wavefunction-level machine-learning models that preserve the formal structure of coupled-cluster theory.
major comments (2)
- [Abstract] Abstract: the central multi-property claim (accurate CC-quality energies, forces, dipoles, quadrupoles, polarizabilities, density, and pair density) is asserted without any numerical error metrics, training-set statistics, loss-function details for the Λ heads, or out-of-distribution test results, rendering the transferability assertion impossible to evaluate from the provided text.
- [Abstract] The weakest load-bearing assumption—that a single model trained solely on localized HF MOs can recover both T and Λ amplitudes with sufficient accuracy and transferability across chemical space without molecule-specific corrections or canonical-orbital baselines—is stated but not accompanied by the ablation or extrapolation tests required to substantiate it.
minor comments (2)
- [Abstract] The notation MōLe-Λ and the description of the Λ1/Λ2 readouts mirroring the T heads would benefit from an explicit equation or diagram showing how the odd sign-equivariant decoding is applied to the left-hand amplitudes.
- [Abstract] The phrase 'substantially expanding the accessible properties' should be accompanied by a direct comparison table (energies/forces vs. full response set) once numerical results are added.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below and indicate the revisions planned for the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central multi-property claim (accurate CC-quality energies, forces, dipoles, quadrupoles, polarizabilities, density, and pair density) is asserted without any numerical error metrics, training-set statistics, loss-function details for the λ heads, or out-of-distribution test results, rendering the transferability assertion impossible to evaluate from the provided text.
Authors: We agree that the abstract would benefit from quantitative support. In the revised manuscript we will add representative error metrics (e.g., MAE for energies, forces, dipoles and polarizabilities), training-set size, and a brief statement on the λ-head loss functions. Out-of-distribution results already appear in Section 4.3; we will add an explicit cross-reference in the abstract. revision: yes
-
Referee: [Abstract] The weakest load-bearing assumption—that a single model trained solely on localized HF MOs can recover both T and λ amplitudes with sufficient accuracy and transferability across chemical space without molecule-specific corrections or canonical-orbital baselines—is stated but not accompanied by the ablation or extrapolation tests required to substantiate it.
Authors: The manuscript already reports size-extensive transferability and out-of-distribution performance on molecules of varying size and composition (Sections 4.2 and 4.4) without molecule-specific corrections. We acknowledge, however, that dedicated ablations versus canonical orbitals are absent. We will add a short discussion of this point and, if space allows, a supplementary ablation table in the revision. revision: partial
Circularity Check
No significant circularity: trained surrogate model on external CCSD data
full rationale
The paper introduces a machine learning model (MōLe-Λ) that learns T and Λ amplitudes from localized HF MOs, trained on external CCSD computations. No equations or steps in the abstract or description reduce the claimed outputs (energies, forces, properties) to fitted inputs by construction or self-definition. The model is presented as a data-driven surrogate with independent validation implied by training on CCSD targets. No load-bearing self-citations or uniqueness theorems are invoked to force the results; the central claims rest on the learned mapping rather than tautological redefinitions. This is a standard non-circular ML surrogate setup.
Axiom & Free-Parameter Ledger
free parameters (1)
- neural network weights
Reference graph
Works this paper leans on
-
[1]
1 Burger, A., Thiede, L., Rønne, N., Bernales, V ., Vijayku- mar, N., Vegge, T., Bhowmik, A., and Aspuru-Guzik, A. Shoot from the hip: Hessian interatomic potentials with- out derivatives.arXiv preprint arXiv:2509.21624, 2025. 1 Chen, Y . and Dral, P. O. One to Rule Them All: A Universal Interatomic Potential Learning across Quan- tum Chemical Levels.Jour...
-
[2]
and G ¨unnemann, S
1 Gao, N. and G ¨unnemann, S. Generalizing Neural Wave Functions. InProceedings of the 40th International Con- ference on Machine Learning, pp. 10708–10726. PMLR, July 2023. URL https://proceedings.mlr. press/v202/gao23c.html. ISSN: 2640-3498. 1 Handy, N. C. and Schaefer III, H. F. On the evaluation of ana- lytic energy derivatives for correlated wave fun...
2023
-
[3]
1 Helgaker, T., Jorgensen, P., and Olsen, J.Molecular electronic-structure theory. John Wiley & Sons, 2013. 1 Jørgensen, P. B. and Bhowmik, A. Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids.npj Computational Materials, 8(1): 183, 2022. 1 Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., ...
-
[4]
2 Kohn, W. and Sham, L. J. Self-consistent equations includ- ing exchange and correlation effects.Physical review, 140(4A):A1133, 1965. 1 Korona, T. and Jeziorski, B. One-electron properties and electrostatic interaction energies from the expectation value expression and wave function of singles and dou- bles coupled cluster theory.The Journal of Chemi- c...
-
[5]
doi: 10.1088/3050-287X/ ae1408
ISSN 3050-287X. doi: 10.1088/3050-287X/ ae1408. URL http://arxiv.org/abs/2508. 21663. arXiv:2508.21663 [cond-mat]. 1 Purvis III, G. D. and Bartlett, R. J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples.The Journal of chemical physics, 76(4):1910– 1918, 1982. 1, 2 Qin, Z., Han, X., Sun, W., Li, D., Kong, L., Barnes,...
-
[6]
1, 3, 3, B.1, B.1 Townsend, J
URL https://openreview.net/forum? id=i5QhkUCzJh. 1, 3, 3, B.1, B.1 Townsend, J. and V ogiatzis, K. D. Data-driven acceleration of the coupled-cluster singles and doubles iterative solver. The journal of physical chemistry letters, 10(14):4129– 4135, 2019. 1 Townsend, J. and V ogiatzis, K. D. Transferable mp2-based machine learning for accurate coupled-clu...
2019
-
[7]
URL https:// doi.org/10.1021/acs.jctc.3c01146
doi: 10.1021/acs.jctc.3c01146. URL https:// doi.org/10.1021/acs.jctc.3c01146. Pub- lisher: American Chemical Society. 1 Venturella, C., Li, J., Hillenbrand, C., Leyva Peralta, X., Liu, J., and Zhu, T. Unified deep learning frame- work for many-body quantum chemistry via Green’s functions.Nature Computational Science, 5(6):502– 513, June 2025. ISSN 2662-84...
-
[8]
S., Feunang, Y
4 Wishart, D. S., Feunang, Y . D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., Sajed, T., Johnson, D., Li, C., Sayeeda, Z., et al. Drugbank 5.0: a major update to the drug- bank database for 2018.Nucleic acids research, 46(D1): D1074–D1082, 2018. B.3 Yu, H., Xu, Z., Qian, X., Qian, X., and Ji, S. Efficient and equivariant graph networks for predicting...
2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.