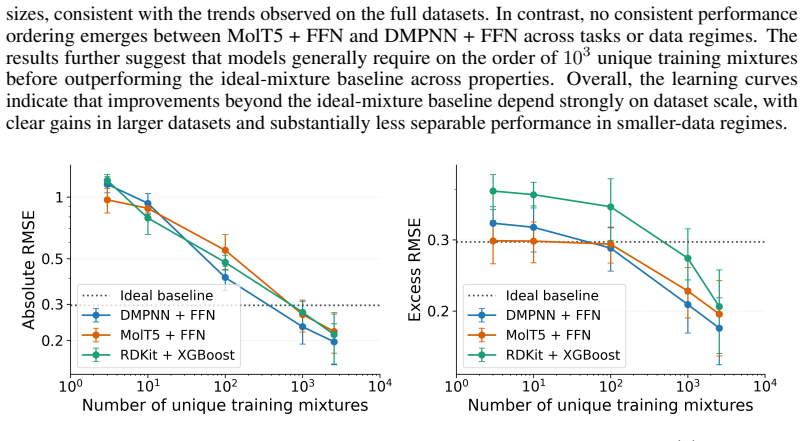
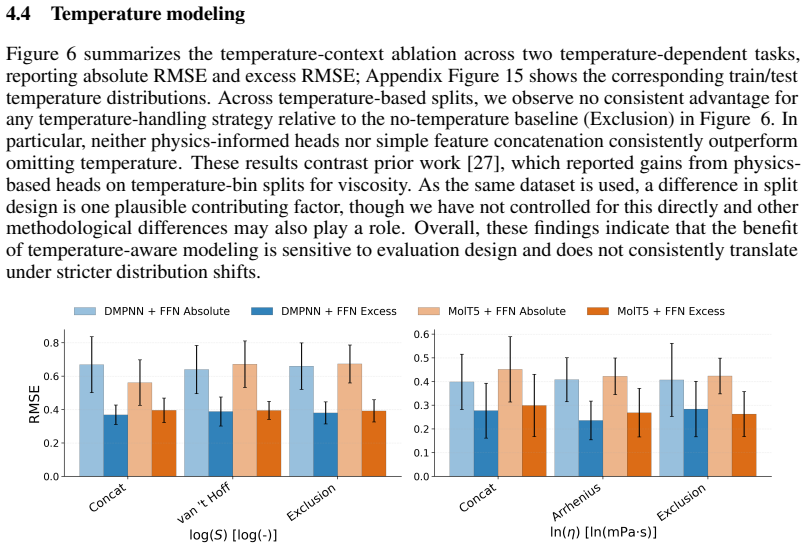
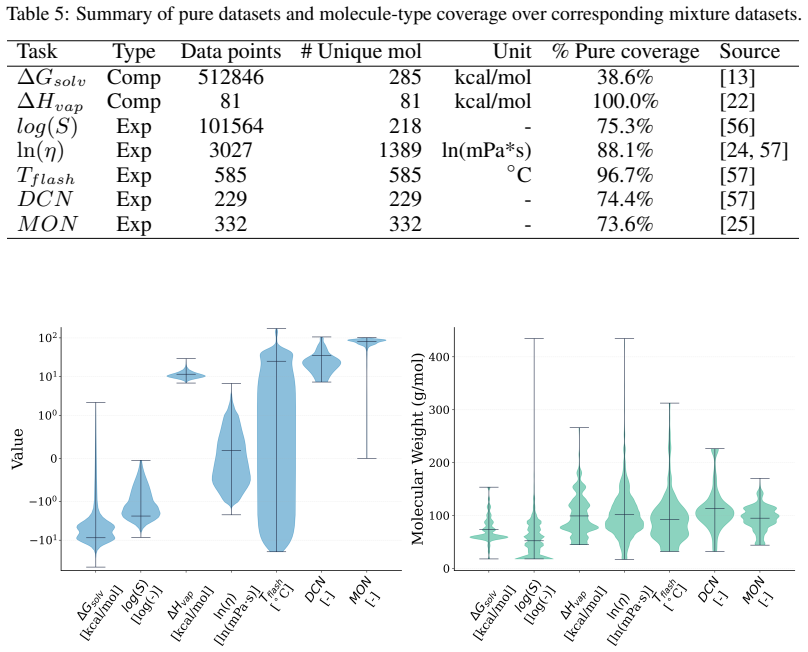
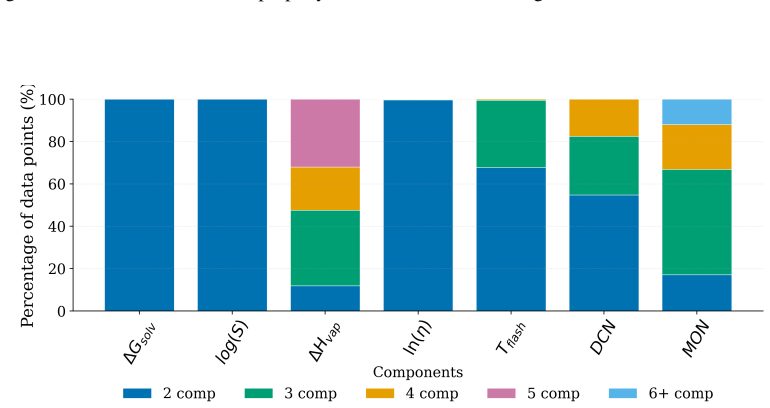
A Systematic Evaluation of Molecular Mixture Behavior Prediction
Pith reviewed 2026-06-29 08:54 UTC · model grok-4.3
The pith
Machine learning models for molecular mixtures can show good overall accuracy while failing to capture non-ideal interactions between components.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
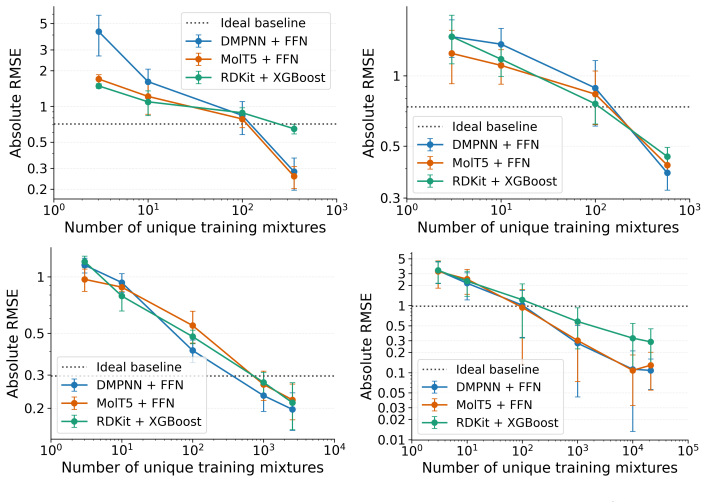
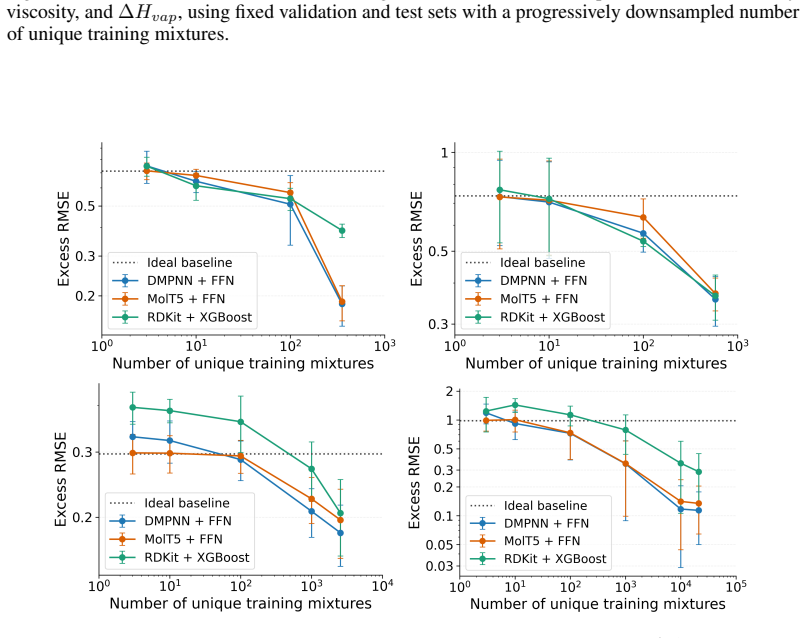
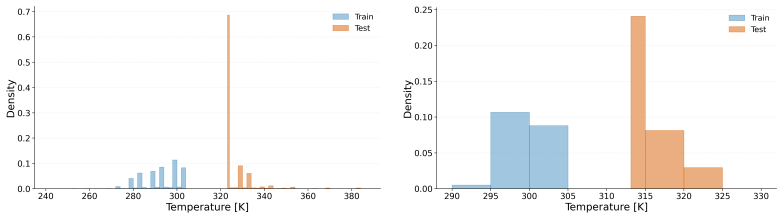
The authors claim that absolute accuracy metrics in mixture property prediction can obscure poor performance on non-ideal components, with substantial drops under molecule-based splits that prevent leakage, identifying transfer to unseen molecules as the main challenge.
What carries the argument
A decomposition framework using ideal-mixture baselines and excess-property metrics to separate pure-compound errors from interaction errors, paired with leakage-aware data splits.
If this is right
- Evaluations of mixture models must include checks on excess properties to verify recovery of non-ideal behavior.
- Performance on strict molecule splits provides a better indicator of generalization than random splits.
- Curated matched datasets of pure and mixture properties enable more reliable benchmarking.
- Models should be assessed for their ability to predict deviations from ideal mixing separately from pure component properties.
Where Pith is reading between the lines
- Explicit modeling of interaction terms could improve recovery of non-ideal effects beyond current end-to-end approaches.
- Similar decomposition techniques might help in evaluating predictions for other complex systems like solutions or alloys.
- The findings imply that larger and more diverse mixture datasets will be necessary to improve transfer performance.
Load-bearing premise
Ideal mixture baselines and excess property calculations isolate non-ideal errors without interference from dataset biases or model assumptions.
What would settle it
Compare model predictions of excess properties on mixtures of entirely new molecules against experimental data to see if the non-ideal component error is low.
Figures
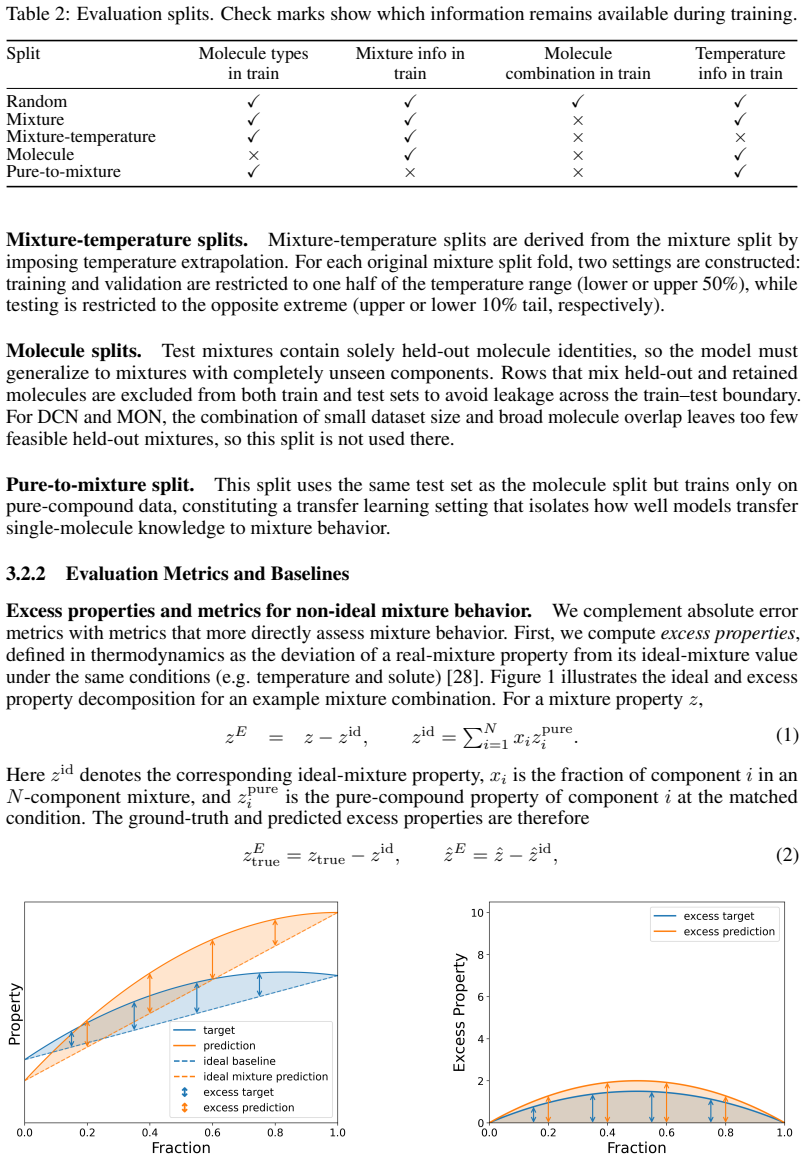
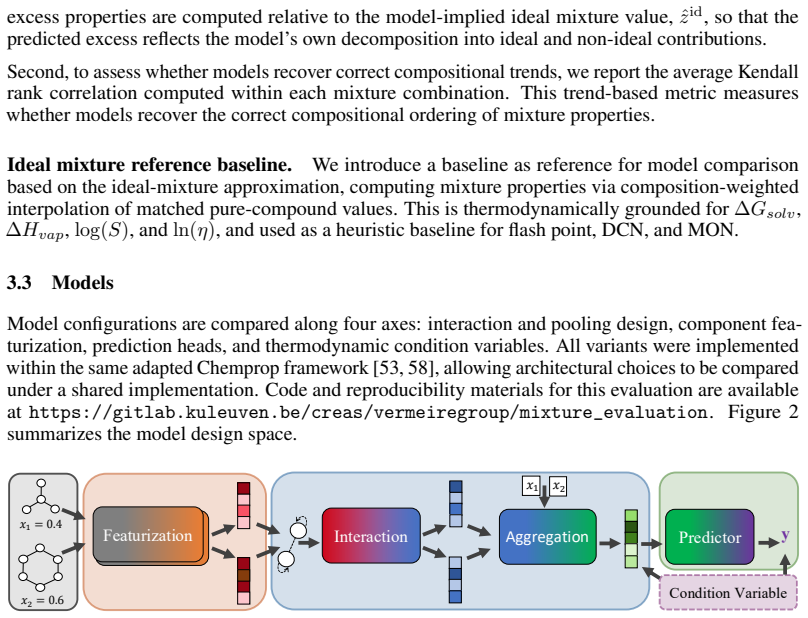
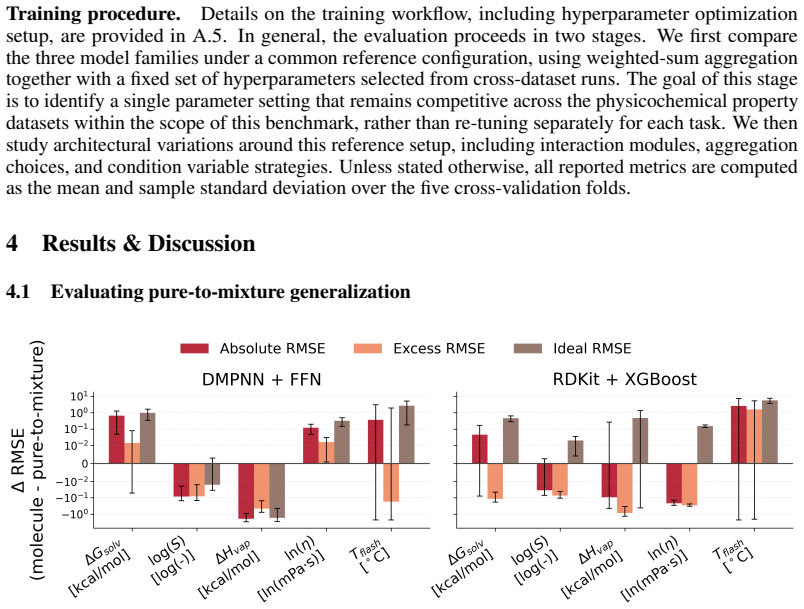
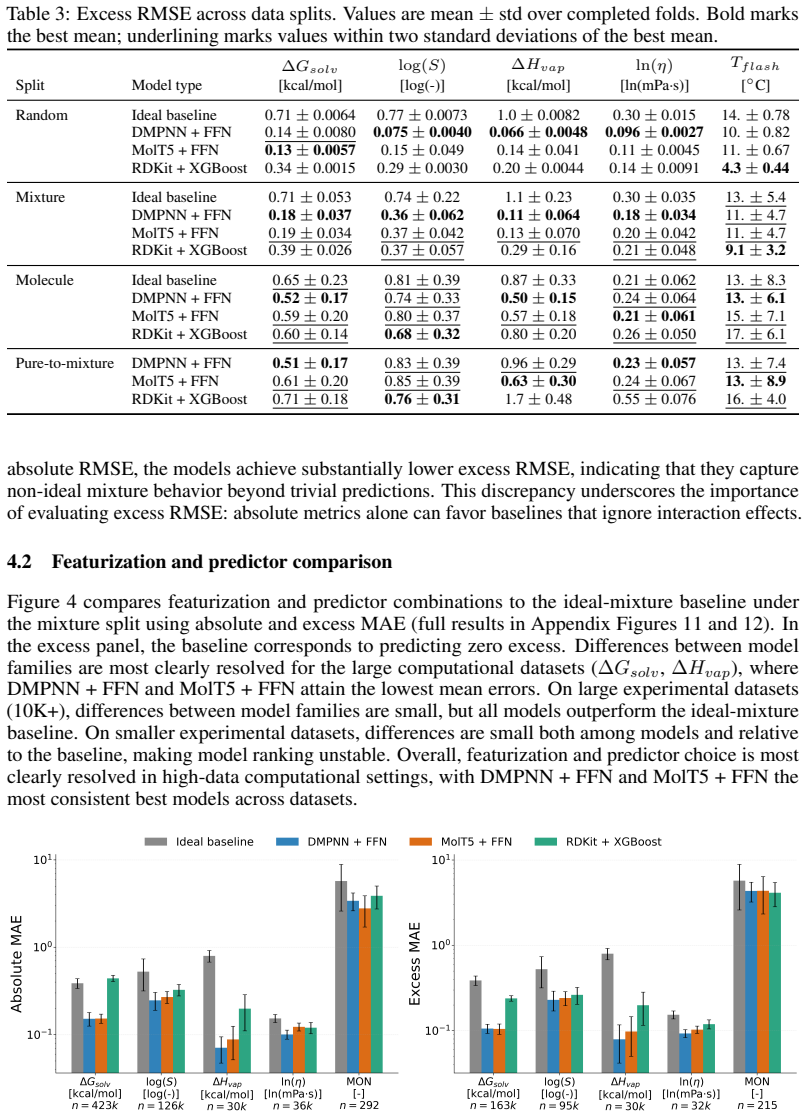
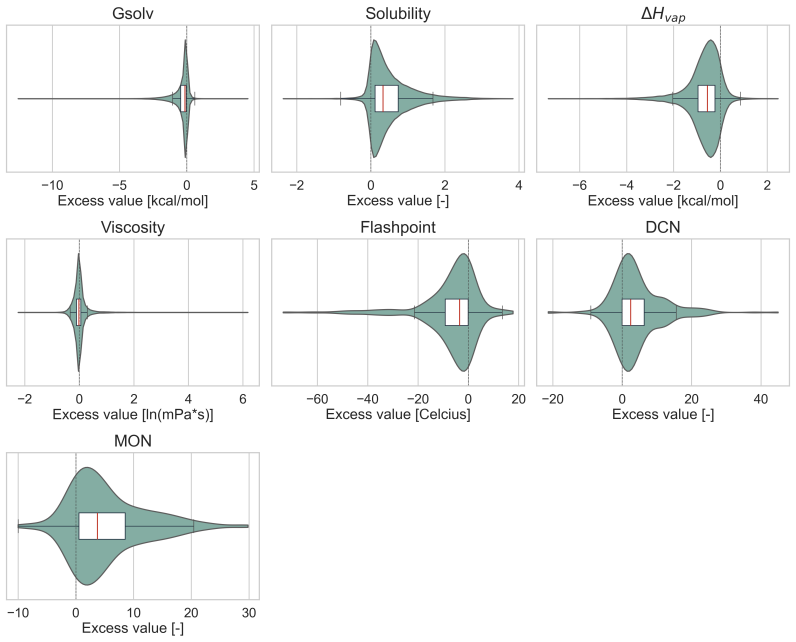
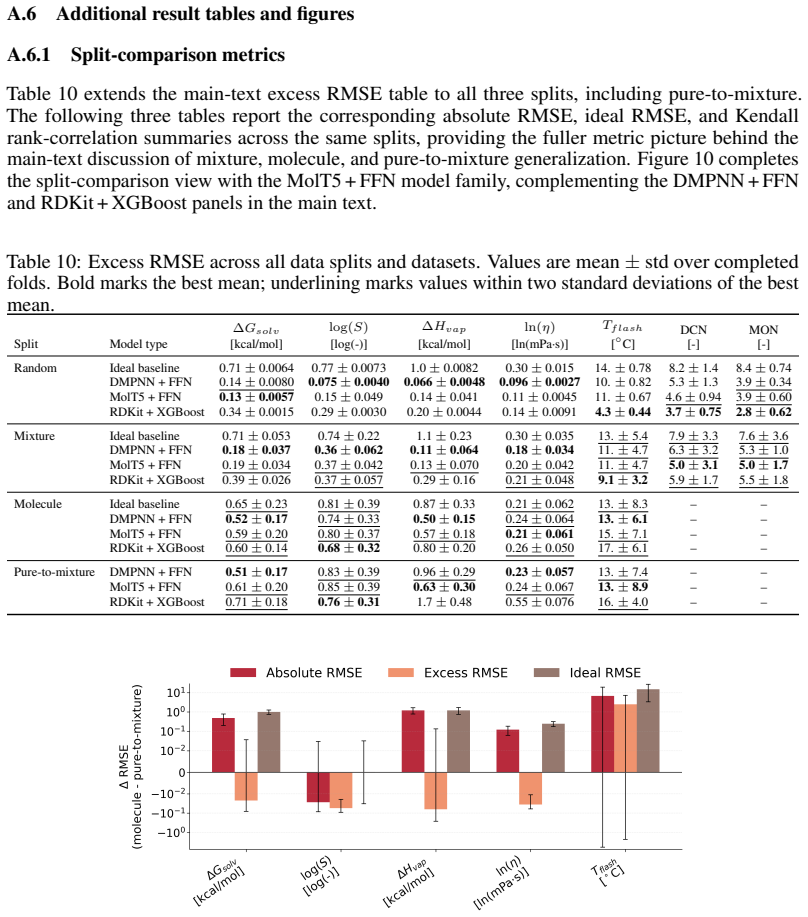
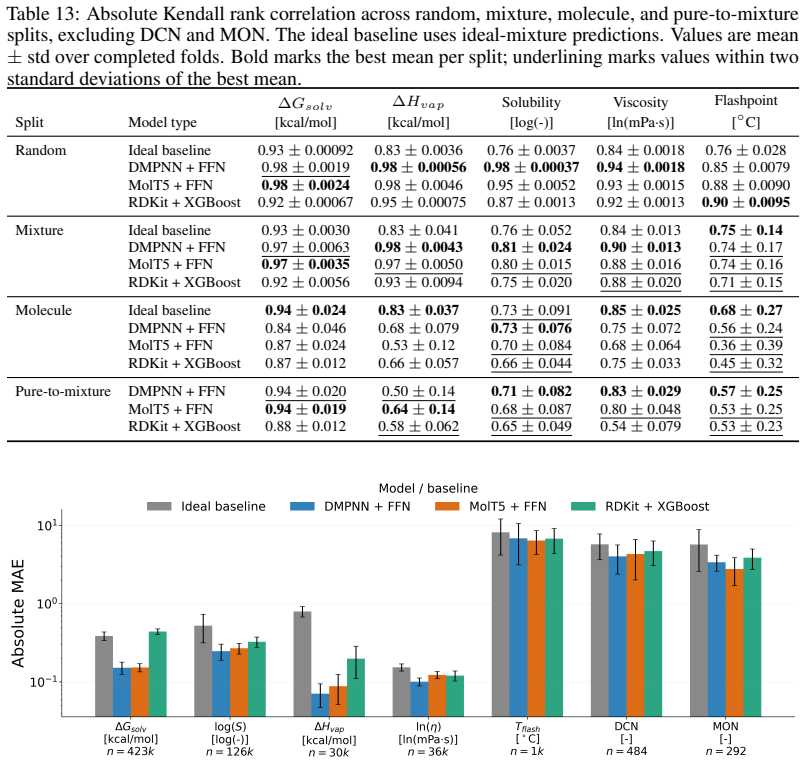
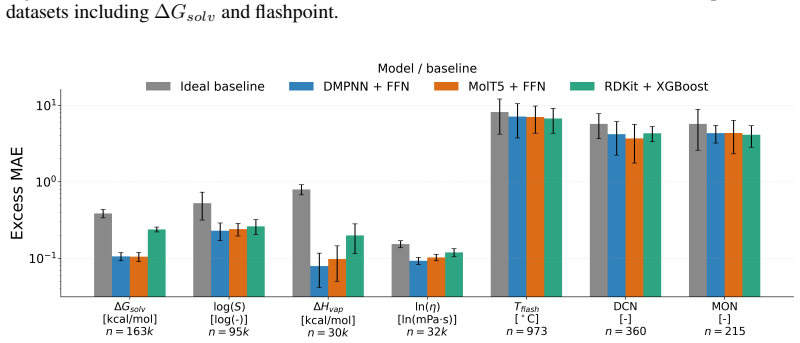
read the original abstract
Machine learning for molecular property prediction has focused largely on pure compounds, even though many practical applications depend on mixtures with intermolecular interactions. Recent work has expanded the availability of mixture datasets, but evaluation still focuses mainly on absolute accuracy. However, absolute errors in mixtures conflate pure-component contributions with deviations from ideal mixing. We propose an evaluation framework that decomposes mixture-property error into pure-compound and interaction (non-ideal) components. The framework combines leakage-aware split protocols, ideal-mixture baselines, and excess-property metrics. To support reproducible benchmarking, we curate seven matched pure and mixture physicochemical property datasets. Across multiple mixture-property tasks and model families, we find that strong absolute accuracy can mask poor recovery of non-ideal mixture behavior, and that performance drops substantially under strict molecule splits. These results identify transfer to unseen molecules as a central challenge in molecular mixture machine learning and motivate evaluation beyond absolute accuracy alone.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an evaluation framework for machine learning models predicting physicochemical properties of molecular mixtures. The framework decomposes prediction error into pure-compound and non-ideal interaction (excess) components via ideal-mixture baselines and excess-property metrics, combined with leakage-aware splits. The authors curate seven matched pure-compound and mixture datasets and evaluate multiple model families, reporting that high absolute accuracy often masks poor recovery of non-ideal behavior while performance drops substantially under strict molecule-based splits. The central conclusion is that transfer to unseen molecules remains a key challenge in molecular mixture machine learning.
Significance. If the decomposition is shown to be free of confounding, the work would be significant for establishing a reproducible benchmarking standard that moves the field beyond absolute-error metrics toward isolating intermolecular interaction effects. The curation of matched datasets and the explicit comparison of absolute vs. excess metrics provide concrete evidence that current models struggle with generalization, which could guide future method development in a practically relevant domain.
minor comments (2)
- The abstract states the decomposition but does not include the explicit equation; adding it (or a reference to the methods section) would improve immediate clarity for readers.
- Figure or table captions should explicitly state the number of molecules in each strict split to allow quick assessment of the generalization gap magnitude.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of our work and the recommendation for minor revision. The report does not enumerate any specific major comments requiring point-by-point rebuttal.
Circularity Check
No circularity: empirical evaluation with independent baselines and metrics
full rationale
The paper is an evaluation study that curates datasets, applies leakage-aware splits, computes ideal-mixture baselines from pure-component data, and uses excess-property metrics to isolate non-ideal contributions. All reported findings (absolute accuracy masking non-ideal recovery, performance drop under molecule splits) are direct empirical observations from running existing model families on these datasets. No derivation, prediction, or uniqueness claim reduces by construction to fitted parameters, self-citations, or ansatzes; the decomposition is a definitional accounting identity using standard thermodynamic excess functions, not a self-referential result. Self-citations, if present, are not load-bearing for the central claims.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Mohamad H. Muhieddine, Shekhar K. Viswanath, Alan Armstrong, Amparo Galindo, and Claire S. Adjiman. Model-based solvent selection for the synthesis and crystallisation of pharmaceutical compounds. Chemical Engineering Science, 264:118125, 2022. DOI: 10.1016/j.ces.2022.118125
-
[2]
Huber, Sara Iborra, and Avelino Corma
George W. Huber, Sara Iborra, and Avelino Corma. Synthesis of Transportation Fuels from Biomass: Chem- istry, Catalysts, and Engineering.Chemical Reviews, 106(9):4044–4098, 2006. DOI: 10.1021/cr068360d
-
[3]
Patel, Absar Lakdawala, Sajan Chourasia, and Rajesh N
Paresh D. Patel, Absar Lakdawala, Sajan Chourasia, and Rajesh N. Patel. Bio fuels for compression ignition engine: A review on engine performance, emission and life cycle analysis.Renewable and Sustainable Energy Reviews, 65:24–43, 2016. DOI: 10.1016/j.rser.2016.06.010
-
[4]
Roger A. Sheldon. The E factor 25 years on: the rise of green chemistry and sustainability.Green Chem., 19:18–43, 2017. DOI: 10.1039/C6GC02157C
-
[5]
David J. C. Constable, Conchita Jimenez-Gonzalez, and Richard K. Henderson. Perspective on Solvent Use in the Pharmaceutical Industry.Organic Process Research & Development, 11(1):133–137, 2007. DOI: 10.1021/op060170h
-
[6]
Clarke, Wei-Chien Tu, Oliver Levers, Andreas Bröhl, and Jason P
Coby J. Clarke, Wei-Chien Tu, Oliver Levers, Andreas Bröhl, and Jason P. Hallett. Green and Sustainable Solvents in Chemical Processes.Chemical Reviews, 118(2):747–800, 2018. DOI: 10.1021/acs.chemrev.7b00571
-
[7]
Zeqing Bao, Gary Tom, Austin Cheng, Alán Aspuru-Guzik, and Christine Allen. Towards the prediction of drug solubility in binary solvent mixtures at various temperatures using machine learning.Research Square, 2024. DOI: 10.21203/rs.3.rs-4170106/v1
-
[8]
Bhupendra Singh Chauhan, Ram Kripal Singh, H.M. Cho, and H.C. Lim. Practice of diesel fuel blends using alternative fuels: A review.Renewable and Sustainable Energy Reviews, 59:1358–1368, 2016. DOI: 10.1016/j.rser.2016.01.062
-
[9]
G. M. Wilson and C. H. Deal. Activity Coefficients and Molecular Structure. Activity Coefficients in Changing Environments-Solutions of Groups.Industrial & Engineering Chemistry Fundamentals, 1(1): 20–23, 1962. DOI: 10.1021/i160001a003
-
[10]
Aage Fredenslund, Russell L. Jones, and John M. Prausnitz. Group-contribution estimation of activity coef- ficients in nonideal liquid mixtures.AIChE Journal, 21(6):1086–1099, 1975. DOI: 10.1002/aic.690210607
-
[11]
Andreas Klamt. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena.The Journal of Physical Chemistry, 99(7):2224–2235, 1995. DOI: 10.1021/j100007a062. 10
-
[12]
Thomas Specht, Mayank Nagda, Sophie Fellenz, Stephan Mandt, Hans Hasse, and Fabian Jirasek. HANNA: hard-constraint neural network for consistent activity coefficient prediction.Chemical Science, 15(47): 19777–19786, 2024. DOI: 10.1039/d4sc05115g
-
[13]
Florence H. Vermeire and William H. Green. Transfer learning for solvation free energies: From quantum chemistry to experiments.Chemical Engineering Journal, 418:129307, 2021. DOI: 10.1016/j.cej.2021.129307
-
[14]
Jan G. Rittig and Alexander Mitsos. Thermodynamics-consistent graph neural networks.Chemical Science, 15:18504–18512, 2024. DOI: 10.1039/D4SC04554H
-
[15]
Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S
Zhenqin Wu, Bharath Ramsundar, Evan N. Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S. Pappu, Karl Leswing, and Vijay Pande. MoleculeNet: a benchmark for molecular machine learning.Chemical Science, 9:513–530, 2018. DOI: 10.1039/C7SC02664A
-
[16]
Kexin Huang, Tianfan Fu, Wenhao Gao, Yue Zhao, Yusuf H Roohani, Jure Leskovec, Connor W. Coley, Cao Xiao, Jimeng Sun, and Marinka Zitnik. Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development. 2021. URLhttps://arxiv.org/abs/2102.09548
-
[17]
Raghunathan Ramakrishnan, Pavlo O. Dral, Matthias Rupp, and O. Anatole von Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules.Scientific Data, 1(1):140022, 2014. DOI: 10.1038/sdata.2014.22
-
[18]
Segler, and Alain C
Nathan Brown, Marco Fiscato, Marwin H.S. Segler, and Alain C. Vaucher. GuacaMol: Benchmarking Models for de Novo Molecular Design.Journal of Chemical Information and Modeling, 59(3):1096–1108,
-
[19]
DOI: 10.1021/acs.jcim.8b00839
-
[20]
Open Graph Benchmark: Datasets for Machine Learning on Graphs
Weihua Hu, Matthias Fey, Marinka Zitnik, Yuxiao Dong, Hongyu Ren, Bowen Liu, Michele Catasta, and Jure Leskovec. Open Graph Benchmark: Datasets for Machine Learning on Graphs. In H. Larochelle, M. Ranzato, R. Hadsell, M.F. Balcan, and H. Lin, editors,Ad- vances in Neural Information Processing Systems, volume 33, pages 22118–22133. Curran Asso- ciates, In...
2020
-
[21]
Sample Efficiency Matters: A Benchmark for Practical Molecular Optimization
Wenhao Gao, Tianfan Fu, Jimeng Sun, and Connor Coley. Sample Efficiency Matters: A Benchmark for Practical Molecular Optimization. In S. Koyejo, S. Mohamed, A. Agarwal, D. Belgrave, K. Cho, and A. Oh, editors,Advances in Neural Information Processing Systems, volume 35, pages 21342–21357. Cur- ran Associates, Inc., 2022. URL https://proceedings.neurips.cc...
-
[22]
Leenhouts, Nathan Morgan, Emad Al Ibrahim, William H
Roel J. Leenhouts, Nathan Morgan, Emad Al Ibrahim, William H. Green, and Florence H. Vermeire. Pooling solvent mixtures for solvation free energy predictions.Chemical Engineering Journal, 513: 162232, 2025. DOI: 10.1016/j.cej.2025.162232
-
[23]
Chew, Mohammad Atif Faiz Afzal, Zachary Kaplan, Eric M
Alex K. Chew, Mohammad Atif Faiz Afzal, Zachary Kaplan, Eric M. Collins, Suraj Gattani, Mayank Misra, Anand Chandrasekaran, Karl Leswing, and Mathew D. Halls. Leveraging high-throughput molecular simulations and machine learning for the design of chemical mixtures.npj Computational Materials, 11 (1):72, 2025. DOI: 10.1038/s41524-025-01552-2
-
[24]
Leenhouts, Tara Larsson, Sebastian Verhelst, and Florence H
Roel J. Leenhouts, Tara Larsson, Sebastian Verhelst, and Florence H. Vermeire. Property prediction of fuel mixtures using pooled graph neural networks.Fuel, 381:133218, 2025. DOI: 10.1016/j.fuel.2024.133218
-
[25]
Camille Bilodeau, Andrei Kazakov, Sukrit Mukhopadhyay, Jillian Emerson, Tom Kalantar, Chris Muzny, and Klavs Jensen. Machine learning for predicting the viscosity of binary liquid mixtures.Chemical Engineering Journal, 464:142454, 2023. DOI: 10.1016/j.cej.2023.142454
-
[26]
Nursulu Kuzhagaliyeva, Samuel Horváth, John Williams, Andre Nicolle, and S. Mani Sarathy. Artificial intelligence-driven design of fuel mixtures.Communications Chemistry, 5(1), 2022. DOI: 10.1038/s42004- 022-00722-3
-
[27]
Dmitry Malikov, Lev Krasnov, Marina Kiseleva, Elizaveta Meshcheriakova, Fedor Kuznetsov, Vladimir Elistratov, Matvei Vasiyarov, Sergei Tatarin, and Stanislav Bezzubov. MixtureSolDB, dataset of solubility values for organic compounds in binary mixtures of solvents at various temperatures.ChemRxiv, 2025. DOI: 10.26434/chemrxiv-2025-m51v8
-
[28]
Mejía-Mendoza, Seyed Mohamad Moosavi, and Benjamin Manuel Sanchez
Ella Miray Rajaonson, Mahyar Rajabi Kochi, Luis M. Mejía-Mendoza, Seyed Mohamad Moosavi, and Benjamin Manuel Sanchez. CheMixHub: Datasets and Benchmarks for Chemical Mixture Property Prediction. InThe Thirty-ninth Annual Conference on Neural Information Processing Systems Datasets and Benchmarks Track, 2025. URLhttps://openreview.net/forum?id=8HUnx0rJNq. 11
2025
-
[29]
J. M. Prausnitz, R. N. Lichtenthaler, and Edmundo Gomes de Azevedo.Molecular thermodynamics of fluid-phase equilibria third edition. Prentice Hall, 1999
1999
-
[30]
Henri Renon and J. M. Prausnitz. Local compositions in thermodynamic excess functions for liquid mixtures.AIChE Journal, 14(1):135–144, 1968. DOI: 10.1002/aic.690140124
-
[31]
Rosana J. Martins, Márcio J. E. de M. Cardoso, and Oswaldo E. Barcia. Excess Gibbs Free Energy Model for Calculating the Viscosity of Binary Liquid Mixtures.Industrial & Engineering Chemistry Research, 39 (3):849–854, 2000. DOI: 10.1021/ie990398b
-
[32]
Yuanbin Liu, Weixiang Hong, and Bingyang Cao. Machine learning for predicting thermodynamic proper- ties of pure fluids and their mixtures.Energy, 188:116091, 2019. DOI: 10.1016/j.energy.2019.116091
-
[33]
Jianyuan Deng, Zhibo Yang, Hehe Wang, Iwao Ojima, Dimitris Samaras, and Fusheng Wang. A systematic study of key elements underlying molecular property prediction.Nature Communications, 14(1), 2023. DOI: 10.1038/s41467-023-41948-6
-
[34]
Roman Joeres, David B. Blumenthal, and Olga V . Kalinina. Data splitting to avoid information leakage with DataSAIL.Nature Communications, 16(1), 2025. DOI: 10.1038/s41467-025-58606-8
-
[35]
Artur M. Schweidtmann, Jan G. Rittig, Jana M. Weber, Martin Grohe, Manuel Dahmen, Kai Leonhard, and Alexander Mitsos. Physical pooling functions in graph neural networks for molecular property prediction. Computers & Chemical Engineering, 172:108202, 2023. DOI: 10.1016/j.compchemeng.2023.108202
-
[36]
Dobbelaere, István Lengyel, Christian V
Maarten R. Dobbelaere, István Lengyel, Christian V . Stevens, and Kevin M. Van Geem. Geometric deep learning for molecular property predictions with chemical accuracy across chemical space.Journal of Cheminformatics, 16(1), 2024. DOI: 10.1186/s13321-024-00895-0
-
[37]
McGill, Florence H
Esther Heid, Charles J. McGill, Florence H. Vermeire, and William H. Green. Characterizing Uncertainty in Machine Learning for Chemistry.Journal of Chemical Information and Modeling, 63(13):4012–4029,
-
[38]
DOI: 10.1021/acs.jcim.3c00373
-
[39]
Shiyi Qin, Shengli Jiang, Jianping Li, Prasanna Balaprakash, Reid C. Van Lehn, and Victor M. Zavala. Capturing molecular interactions in graph neural networks: a case study in multi-component phase equilibrium.Digital Discovery, 2:138–151, 2023. DOI: 10.1039/D2DD00045H
-
[40]
Di Wu, Zutao Zhu, Jun Zhang, Huaqiang Wen, Saimeng Jin, and Weifeng Shen. An Interpretable Solute–Solvent Interactive Attention Module Intensified Graph-Learning Architecture toward Enhancing the Prediction Accuracy of an Infinite Dilution Activity Coefficient.Industrial & Engineering Chemistry Research, 63(19):8741–8750, 2024. DOI: 10.1021/acs.iecr.4c00107
-
[41]
Benedikt Winter, Clemens Winter, Johannes Schilling, and André Bardow. A smile is all you need: predicting limiting activity coefficients from SMILES with natural language processing.Digital Discovery, 1:859–869, 2022. DOI: 10.1039/D2DD00058J
-
[42]
Molecular machine learning in chemical process design.Current Opinion in Chemical Engineering, 52:101239, 2026
Jan G Rittig, Manuel Dahmen, Martin Grohe, Philippe Schwaller, and Alexander Mitsos. Molecular machine learning in chemical process design.Current Opinion in Chemical Engineering, 52:101239, 2026
2026
-
[43]
Jan G. Rittig, Kobi C. Felton, Alexei A. Lapkin, and Alexander Mitsos. Gibbs–Duhem-informed neu- ral networks for binary activity coefficient prediction.Digital Discovery, 2:1752–1767, 2023. DOI: 10.1039/D3DD00103B
-
[44]
Hengrui Zhang, Tianxing Lai, Jie Chen, Arumugam Manthiram, James M. Rondinelli, and Wei Chen. Learning Molecular Mixture Property Using Chemistry-Aware Graph Neural Network.PRX Energy, 3: 023006, 2024. DOI: 10.1103/PRXEnergy.3.023006
-
[45]
Ulderico Di Caprio, Florence Vermeire, Tom Van Gerven, and M. Enis Leblebici. Physics-informed machine learning predicting CO2 capture performances of organic mixtures.Chemical Engineering and Processing - Process Intensification, 216:110410, 2025. DOI: 10.1016/j.cep.2025.110410
-
[46]
Rittig, Elie Akanny, Sandip Bhattacharya, Christina Kohlmann, and Alexander Mitsos
Christoforos Brozos, Jan G. Rittig, Elie Akanny, Sandip Bhattacharya, Christina Kohlmann, and Alexander Mitsos. Predicting the temperature-dependent CMC of surfactant mixtures with graph neural networks. Computers & Chemical Engineering, 198:109085, 2025. DOI: 10.1016/j.compchemeng.2025.109085
-
[47]
Deep Neural Networks for Multicomponent Molecular Systems.ACS Omega, 5(33): 21042–21053, 2020
Kyohei Hanaoka. Deep Neural Networks for Multicomponent Molecular Systems.ACS Omega, 5(33): 21042–21053, 2020. DOI: 10.1021/acsomega.0c02599
-
[48]
BP-Kelley/descriptastorus: Descriptor computation(chemistry) and (optional) storage for Machine Learning, 2026
Bp-Kelley. BP-Kelley/descriptastorus: Descriptor computation(chemistry) and (optional) storage for Machine Learning, 2026. URL https://github.com/bp-kelley/descriptastorus. Accessed: May 4, 2026. 12
2026
-
[49]
Mordred: a molecular descriptor calculator.Journal of Cheminformatics, 10(1):4, 2018
Hirotomo Moriwaki, Yu-Shi Tian, Norihito Kawashita, and Tatsuya Takagi. Mordred: a molecular descriptor calculator.Journal of Cheminformatics, 10(1):4, 2018. DOI: 10.1186/s13321-018-0258-y
-
[50]
Extended-connectivity fingerprints.Journal of Chemical Information and Modeling, 50(5):742–754, 2010
David Rogers and Mathew Hahn. Extended-Connectivity Fingerprints.Journal of Chemical Information and Modeling, 50(5):742–754, 2010. DOI: 10.1021/ci100050t
-
[51]
Translation be- tween Molecules and Natural Language
Carl Edwards, Tuan Lai, Kevin Ros, Garrett Honke, Kyunghyun Cho, and Heng Ji. Translation be- tween Molecules and Natural Language. In Yoav Goldberg, Zornitsa Kozareva, and Yue Zhang, editors, Proceedings of the 2022 Conference on Empirical Methods in Natural Language Processing, pages 375–413, Abu Dhabi, United Arab Emirates, 2022. Association for Comput...
-
[52]
Juncai Li and Xiaofei Jiang. Mol-BERT: An Effective Molecular Representation with BERT for Molecular Property Prediction.Wireless Communications and Mobile Computing, 2021(1):7181815, 2021. DOI: 10.1155/2021/7181815
-
[53]
Kevin Yang, Kyle Swanson, Wengong Jin, Connor Coley, Philipp Eiden, Hua Gao, Angel Guzman-Perez, Timothy Hopper, Brian Kelley, Miriam Mathea, Andrew Palmer, V olker Settels, Tommi Jaakkola, Klavs Jensen, and Regina Barzilay. Analyzing Learned Molecular Representations for Property Prediction. Journal of Chemical Information and Modeling, 59(8):3370–3388, ...
-
[54]
Schoenholz, Patrick F
Justin Gilmer, Samuel S. Schoenholz, Patrick F. Riley, Oriol Vinyals, and George E. Dahl. Neural Message Passing for Quantum Chemistry. In Doina Precup and Yee Whye Teh, editors,Proceedings of the 34th International Conference on Machine Learning, volume 70 ofProceedings of Machine Learning Research, pages 1263–1272. PMLR, 2017. URLhttps://proceedings.mlr...
2017
-
[55]
Greenman, Yunsie Chung, Shih-Cheng Li, David E
Esther Heid, Kevin P. Greenman, Yunsie Chung, Shih-Cheng Li, David E. Graff, Florence H. Vermeire, Haoyang Wu, William H. Green, and Charles J. McGill. Chemprop: A Machine Learning Package for Chemical Property Prediction.Journal of Chemical Information and Modeling, 64(1):9–17, 2024. DOI: 10.1021/acs.jcim.3c01250
-
[56]
Fabian Jirasek and Hans Hasse. Combining Machine Learning with Physical Knowledge in Thermodynamic Modeling of Fluid Mixtures.Annual Review of Chemical and Biomolecular Engineering, 14(V olume 14, 2023):31–51, 2023. DOI: 10.1146/annurev-chembioeng-092220-025342
-
[57]
Edgar Ivan Sanchez Medina, Steffen Linke, Martin Stoll, and Kai Sundmacher. Gibbs–Helmholtz graph neural network: capturing the temperature dependency of activity coefficients at infinite dilution.Digital Discovery, 2:781–798, 2023. DOI: 10.1039/D2DD00142J
-
[58]
Lev Krasnov, Dmitry Malikov, Marina Kiseleva, Sergei Tatarin, Sergey Sosnin, and Stanislav Bezzubov. BigSolDB 2.0, dataset of solubility values for organic compounds in different solvents at various tempera- tures.Scientific Data, 12(1):1236, 2025. DOI: 10.1038/s41597-025-05559-8
-
[59]
Tara Larsson, Florence Vermeire, and Sebastian Verhelst. Machine learning for fuel property predictions: A multi-task and transfer learning approach.SAE Technical Paper Series, 1, 2023. DOI: 10.4271/2023-01- 0337
-
[60]
Graff, Nathan K
David E. Graff, Nathan K. Morgan, Jackson W. Burns, Anna C. Doner, Brian Li, Shih-Cheng Li, Joel Manu, Angiras Menon, Hao-Wei Pang, Haoyang Wu, Akshat Shirish Zalte, Jonathan W. Zheng, Connor W. Coley, William H. Green, and Kevin P. Greenman. Chemprop v2: An Efficient, Modular Machine Learning Package for Chemical Property Prediction.Journal of Chemical I...
-
[61]
DOI: 10.1021/acs.jcim.5c02332
-
[62]
Emmanuel Noutahi, Cas Wognum, Hadrien Mary, Honoré Hounwanou, Kyle M. Kovary, Desmond Gilmour, thibaultvarin r, Jackson Burns, Julien St-Laurent, t, DomInvivo, Saurav Maheshkar, and rbyrne momatx. datamol-io/molfeat: 0.9.4, 2023. URLhttps://doi.org/10.5281/zenodo.8373019
-
[63]
RDKit: Open-source cheminformatics., 2026
Greg Landrum. RDKit: Open-source cheminformatics., 2026. URL https://www.rdkit.org/. Ac- cessed: May 4, 2026
2026
-
[64]
Deep Sets
Manzil Zaheer, Satwik Kottur, Siamak Ravanbakhsh, Barnabas Poczos, Ruslan Salakhutdinov, and Alexander Smola. Deep Sets. InAdvances in Neural Information Processing Sys- tems 30, 2017. URL https://proceedings.neurips.cc/paper_files/paper/2017/file/ f22e4747da1aa27e363d86d40ff442fe-Paper.pdf
2017
-
[65]
Cicero dos Santos, Bing Xiang, and Bowen Zhou. Attentive Pooling Networks.arXiv preprint arXiv:1602.03609, 2016. URLhttps://arxiv.org/abs/1602.03609. 13
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[66]
Order Matters: Sequence to sequence for sets
Oriol Vinyals, Samy Bengio, and Manjunath Kudlur. Order Matters: Sequence to Sequence for Sets.arXiv preprint arXiv:1511.06391, 2016. URLhttps://arxiv.org/abs/1511.06391
work page internal anchor Pith review Pith/arXiv arXiv 2016
-
[67]
Tianqi Chen and Carlos Guestrin. XGBoost: A Scalable Tree Boosting System. InProceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’16, page 785–794, New York, NY , USA, 2016. Association for Computing Machinery. DOI: 10.1145/2939672.2939785
-
[68]
Massively Parallel Hyperparameter Tuning.CoRR, abs/1810.05934, 2018
Liam Li, Kevin Jamieson, Afshin Rostamizadeh, Ekaterina Gonina, Moritz Hardt, Benjamin Recht, and Ameet Talwalkar. Massively Parallel Hyperparameter Tuning.CoRR, abs/1810.05934, 2018. URL http://arxiv.org/abs/1810.05934
-
[69]
Tune: A Research Platform for Distributed Model Selection and Training
Richard Liaw, Eric Liang, Robert Nishihara, Philipp Moritz, Joseph E Gonzalez, and Ion Stoica. Tune: A Research Platform for Distributed Model Selection and Training.arXiv preprint arXiv:1807.05118, 2018. A Technical Appendices and Supplementary Material This appendix collects the implementation and data details that support the main evaluation. It first ...
work page internal anchor Pith review Pith/arXiv arXiv 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.