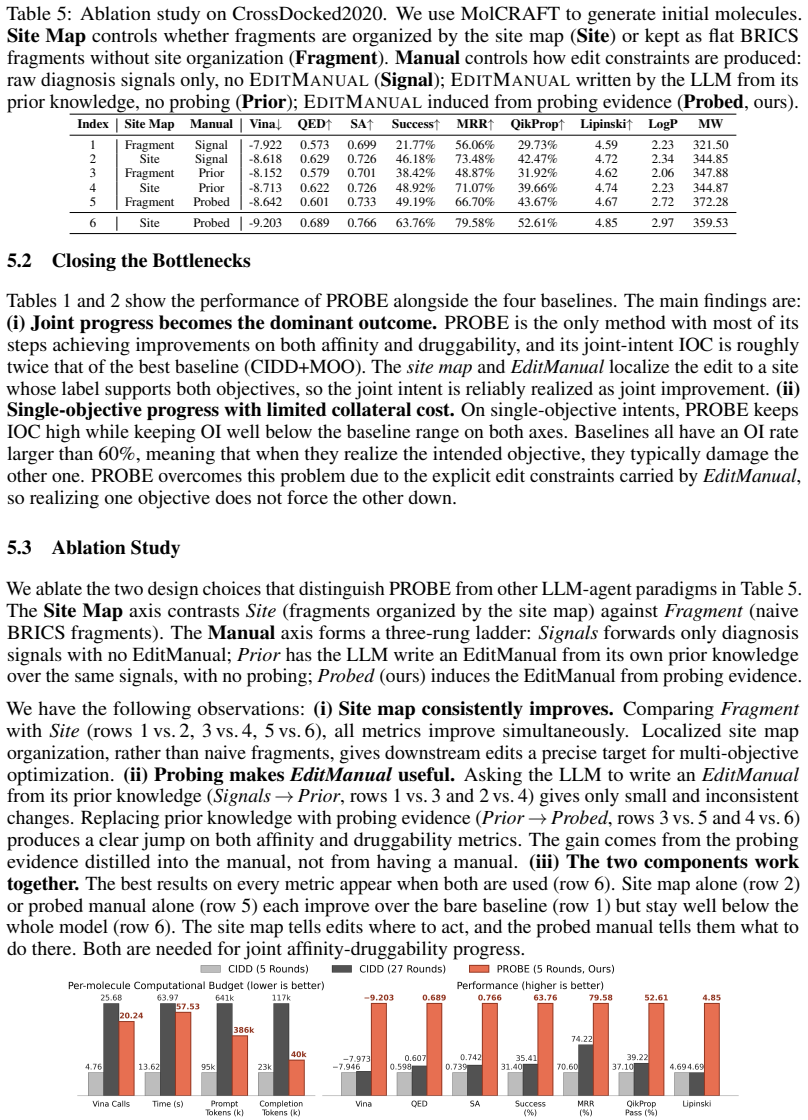
Probe Before You Edit: Probing-Guided Molecular Optimization for LLM Agents in Structure-Based Drug Design
Pith reviewed 2026-06-28 18:58 UTC · model grok-4.3
The pith
PROBE uses controlled probe edits to build site maps that guide LLM agents toward edits improving both binding affinity and druggability.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
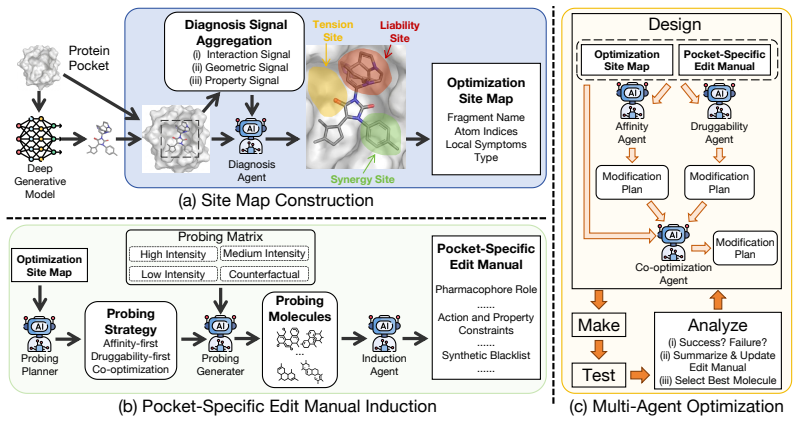
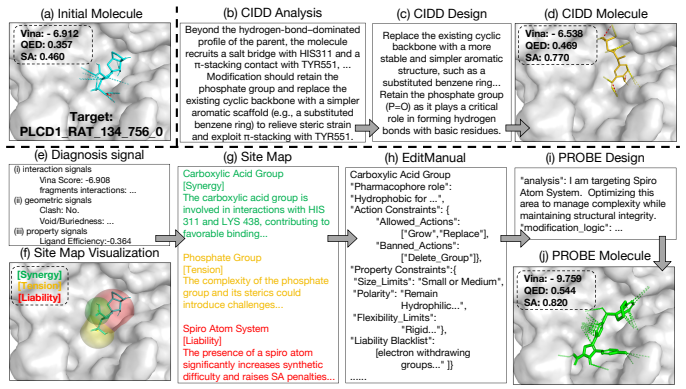
The central claim is that edit-response probing first decomposes the ligand into editable sites and constructs a pocket-specific site map that flags where joint affinity-druggability gains are plausible, where the objectives are likely in tension, and where liability substructures should change; the responses are then distilled into an EditManual that, together with the map, directs an iterative multi-agent loop of affinity, druggability, and co-optimization agents to generate edits that more frequently satisfy both objectives simultaneously.
What carries the argument
edit-response probing that builds a pocket-specific site map and EditManual from controlled edits on editable sites to guide subsequent optimization
If this is right
- Single molecular edits improve both binding affinity and druggability together more often than in standard LLM pipelines.
- Cases in which a gain on one objective produces a loss on the other become less frequent.
- The method attains state-of-the-art performance on the CrossDocked2020 benchmark.
- The diagnostic failure modes of unguided editing are substantially reduced.
Where Pith is reading between the lines
- The same probing step could be inserted into other multi-objective molecular design loops that pit affinity against selectivity or ADMET properties.
- Site-map construction may reduce the total number of LLM calls needed by pruning low-value edit directions early.
- The approach invites testing whether the probe-derived guidance transfers across protein families without retraining the underlying language model.
Load-bearing premise
Responses from a limited set of controlled probe edits on editable sites will generalize to predict joint affinity-druggability outcomes for later optimization edits without systematic bias from probe selection or LLM prompting.
What would settle it
A controlled experiment on held-out ligands in which the frequency of joint-improvement edits remains unchanged when the site map and EditManual are removed from the agent loop would falsify the claim that probing enables better joint optimization.
Figures
read the original abstract
Structure-based drug design increasingly employs LLM agents to iteratively refine ligands against a target pocket, yet a viable ligand must satisfy two often-conflicting objectives -- binding affinity and druggability -- which single optimization steps rarely improve together. To quantify this difficulty, we introduce two diagnostic metrics: the first measures how often a single edit improves both objectives, and the second measures how often a gain on one objective comes with a loss on the other. Applying these diagnostics to current LLM-agent pipelines exposes a consistent failure mode: the agent performs molecular editing without knowing how the pocket-ligand complex responds to local modifications, thus rarely achieving joint improvement. Inspired by medicinal chemists, who probe the pocket-ligand complex with controlled analog edits before choosing an optimization direction, we propose \textbf{PROBE}, an optimization framework built around edit-response probing. PROBE first decomposes the ligand into editable sites and builds a pocket-specific \textbf{site map} that flags where joint gains are plausible, where the two objectives are likely in tension, and where liability substructures should be changed; it then performs controlled probe edits whose responses are distilled into an \textbf{EditManual}. Guided by the site map and EditManual, PROBE runs an iterative multi-agent loop in which an affinity agent, a druggability agent, and a co-optimization agent jointly produce edits. On the CrossDocked2020 benchmark, PROBE achieves state-of-the-art performance and substantially mitigates the failure modes exposed by our diagnostics metrics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that LLM agents for iterative ligand refinement in structure-based drug design rarely achieve simultaneous gains in binding affinity and druggability. It introduces two diagnostic metrics to quantify this failure mode, proposes the PROBE framework that decomposes ligands into editable sites, constructs a pocket-specific site map, performs controlled probe edits to build an EditManual, and then runs a guided multi-agent optimization loop (affinity, druggability, and co-optimization agents). On the CrossDocked2020 benchmark, PROBE is reported to reach state-of-the-art performance while substantially reducing the diagnosed failure modes.
Significance. If the empirical results and generalization hold, the work offers a concrete, benchmark-driven method to mitigate a recurring joint-optimization failure in LLM-based molecular design by emulating medicinal-chemistry probing. The independent definition of the diagnostic metrics (separate from the optimization loop) is a methodological strength, as is the explicit focus on falsifiable failure-mode quantification rather than post-hoc success rates alone.
major comments (2)
- [Abstract and §4] Abstract and §4 (Results): the central claim that PROBE 'substantially mitigates the failure modes' and achieves SOTA rests on the assumption that responses from a limited set of controlled probe edits on pre-identified editable sites generalize to predict joint affinity-druggability outcomes for subsequent multi-agent edits. No ablation or sensitivity analysis is presented that varies probe-site decomposition heuristics or LLM prompting strategies to test for systematic bias in the site map or EditManual.
- [§3.2] §3.2 (PROBE framework): the site map flags (joint-gain plausible, tension, liability) are derived from probe responses, yet the manuscript provides no direct validation that these flags remain predictive when the probe edits are replaced by the actual optimization edits produced by the multi-agent loop; this is load-bearing for the claim that probing reliably guides co-optimization.
minor comments (2)
- [Table captions and §4] Table captions and §4: exact definitions of the two diagnostic metrics (joint-improvement rate and trade-off rate), the precise baselines, data splits, and statistical tests used for the CrossDocked2020 comparison are not summarized in the abstract and should be stated explicitly in the results tables.
- [Methods] Notation: the terms 'site map' and 'EditManual' are used throughout without a concise formal definition or pseudocode in the methods section; a one-paragraph boxed definition would improve clarity.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback. The comments highlight important aspects of validation that can strengthen the manuscript. We address each major comment below and commit to incorporating additional analyses in the revision.
read point-by-point responses
-
Referee: [Abstract and §4] Abstract and §4 (Results): the central claim that PROBE 'substantially mitigates the failure modes' and achieves SOTA rests on the assumption that responses from a limited set of controlled probe edits on pre-identified editable sites generalize to predict joint affinity-druggability outcomes for subsequent multi-agent edits. No ablation or sensitivity analysis is presented that varies probe-site decomposition heuristics or LLM prompting strategies to test for systematic bias in the site map or EditManual.
Authors: We agree that the generalization assumption is central and that explicit sensitivity analyses would provide stronger support. The current CrossDocked2020 results demonstrate mitigation of the diagnosed failure modes, but to directly test for bias we will add, in the revised manuscript, ablations that vary (i) the ligand decomposition heuristics used to identify editable sites and (ii) the prompting strategies employed during probing. These will quantify any systematic effects on site-map construction and downstream performance. revision: yes
-
Referee: [§3.2] §3.2 (PROBE framework): the site map flags (joint-gain plausible, tension, liability) are derived from probe responses, yet the manuscript provides no direct validation that these flags remain predictive when the probe edits are replaced by the actual optimization edits produced by the multi-agent loop; this is load-bearing for the claim that probing reliably guides co-optimization.
Authors: The site-map flags are designed to be predictive on the basis of controlled probe responses, and the multi-agent loop is guided by them. We acknowledge that a direct head-to-head comparison between probe-derived flags and the outcomes of the subsequent multi-agent edits is absent. In revision we will add an analysis that extracts the flags applied during optimization runs and measures their predictive accuracy against the observed affinity-druggability changes, thereby validating the load-bearing assumption. revision: yes
Circularity Check
No significant circularity; empirical framework evaluated on external benchmark
full rationale
The paper defines two diagnostic metrics independently to quantify joint improvement failures, constructs the PROBE site map and EditManual from probe responses as a methodological step, and reports performance on the external CrossDocked2020 benchmark. No equations, fitted parameters renamed as predictions, or self-citation chains appear in the provided text that would reduce any claimed result to its own inputs by construction. The derivation chain is self-contained against the benchmark evaluation.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption LLM agents can be prompted to perform controlled molecular edits and interpret their effects on affinity and druggability
invented entities (2)
-
site map
no independent evidence
-
EditManual
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Ligand efficiency indices as guideposts for drug discovery.Drug discovery today, 10(7):464–469, 2005
Cele Abad-Zapatero and James T Metz. Ligand efficiency indices as guideposts for drug discovery.Drug discovery today, 10(7):464–469, 2005
2005
-
[2]
Liddia: Language-based intelligent drug discovery agent
Reza Averly, Frazier N Baker, Ian A Watson, and Xia Ning. Liddia: Language-based intelligent drug discovery agent. InProceedings of the 2025 Conference on Empirical Methods in Natural Language Processing, pages 12015–12039, 2025
2025
-
[3]
New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays
Jonathan B Baell and Georgina A Holloway. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. Journal of medicinal chemistry, 53(7):2719–2740, 2010
2010
-
[4]
The first general index of molecular complexity.Journal of the American Chemical Society, 103(12):3599–3601, 1981
Steven H Bertz. The first general index of molecular complexity.Journal of the American Chemical Society, 103(12):3599–3601, 1981
1981
-
[5]
Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
G Richard Bickerton, Gaia V Paolini, Jérémy Besnard, Sorel Muresan, and Andrew L Hopkins. Quantifying the chemical beauty of drugs.Nature chemistry, 4(2):90–98, 2012
2012
-
[6]
Generating novel leads for drug discovery using llms with logical feedback
Shreyas Bhat Brahmavar, Ashwin Srinivasan, Tirtharaj Dash, Sowmya Ramaswamy Krishnan, Lovekesh Vig, Arijit Roy, and Raviprasad Aduri. Generating novel leads for drug discovery using llms with logical feedback. InProceedings of the AAAI conference on artificial intelligence, volume 38, pages 21–29, 2024
2024
-
[7]
Decomposed direct preference optimization for structure-based drug design.Transactions on Machine Learning Research
Xiwei Cheng, Xiangxin Zhou, Yuwei Yang, Yu Bao, and Quanquan Gu. Decomposed direct preference optimization for structure-based drug design.Transactions on Machine Learning Research
-
[8]
On the art of compiling and using’drug-like’chemical fragment spaces.ChemMedChem, 3(10):1503, 2008
Jorg Degen, Christof Wegscheid-Gerlach, Andrea Zaliani, and Matthias Rarey. On the art of compiling and using’drug-like’chemical fragment spaces.ChemMedChem, 3(10):1503, 2008
2008
-
[9]
Tagmol: Target-aware gradient-guided molecule generation
Vineeth Dorna, D Subhalingam, Keshav Kolluru, Shreshth Tuli, Mrityunjay Singh, Saurabh Singal, NM Anoop Krishnan, and Sayan Ranu. Tagmol: Target-aware gradient-guided molecule generation. InICML’24 Workshop ML for Life and Material Science: From Theory to Industry Applications
-
[10]
Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions.Journal of cheminfor- matics, 1(1):8, 2009
Peter Ertl and Ansgar Schuffenhauer. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions.Journal of cheminfor- matics, 1(1):8, 2009
2009
-
[11]
Three-dimensional convolutional neural networks and a cross- docked data set for structure-based drug design.Journal of chemical information and modeling, 60(9):4200–4215, 2020
Paul G Francoeur, Tomohide Masuda, Jocelyn Sunseri, Andrew Jia, Richard B Iovanisci, Ian Snyder, and David R Koes. Three-dimensional convolutional neural networks and a cross- docked data set for structure-based drug design.Journal of chemical information and modeling, 60(9):4200–4215, 2020
2020
-
[12]
Cidd: Collaborative intelligence for structure-based drug design empowered by llms
Bowen Gao, Yanwen Huang, Yiqiao Liu, Wenxuan Xie, Bowei He, Haichuan Tan, Wei-Ying Ma, Ya-Qin Zhang, and Yanyan Lan. Cidd: Collaborative intelligence for structure-based drug design empowered by llms. InThe Thirty-ninth Annual Conference on Neural Information Processing Systems, 2025
2025
-
[13]
Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges.Tetrahedron, 36(22):3219–3228, 1980
Johann Gasteiger and Mario Marsili. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges.Tetrahedron, 36(22):3219–3228, 1980
1980
-
[14]
A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery
Arup K Ghose, Vellarkad N Viswanadhan, and John J Wendoloski. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. a qualitative and quantitative characterization of known drug databases.Journal of combinatorial chemistry, 1(1):55–68, 1999
1999
-
[15]
3d equivariant diffusion for target-aware molecule generation and affinity prediction
Jiaqi Guan, Wesley Wei Qian, Xingang Peng, Yufeng Su, Jian Peng, and Jianzhu Ma. 3d equivariant diffusion for target-aware molecule generation and affinity prediction. In The Eleventh International Conference on Learning Representations, 2023. URL https: //openreview.net/forum?id=kJqXEPXMsE0. 10
2023
-
[16]
Decompdiff: Diffusion models with decomposed priors for structure- based drug design
Jiaqi Guan, Xiangxin Zhou, Yuwei Yang, Yu Bao, Jian Peng, Jianzhu Ma, Qiang Liu, Liang Wang, and Quanquan Gu. Decompdiff: Diffusion models with decomposed priors for structure- based drug design. InInternational Conference on Machine Learning, pages 11827–11846. PMLR, 2023
2023
-
[17]
The hypervolume indicator: Computational problems and algorithms.ACM Computing Surveys (CSUR), 54(6):1–42, 2021
Andreia P Guerreiro, Carlos M Fonseca, and Luís Paquete. The hypervolume indicator: Computational problems and algorithms.ACM Computing Surveys (CSUR), 54(6):1–42, 2021
2021
-
[18]
arXiv preprint arXiv:2308.07413 , year=
Charles Harris, Kieran Didi, Arian R Jamasb, Chaitanya K Joshi, Simon V Mathis, Pietro Lio, and Tom Blundell. Benchmarking generated poses: How rational is structure-based drug design with generative models?arXiv preprint arXiv:2308.07413, 2023
-
[19]
Empowering llms for structure-based drug design via exploration-augmented latent inference
Xuanning Hu, Anchen Li, Qianli Xing, Jinglong Ji, Hao Tuo, and Bo Yang. Empowering llms for structure-based drug design via exploration-augmented latent inference. InProceedings of the ACM Web Conference 2026, pages 4244–4255, 2026
2026
-
[20]
Protein-ligand interaction prior for binding- aware 3d molecule diffusion models
Zhilin Huang, Ling Yang, Xiangxin Zhou, Zhilong Zhang, Wentao Zhang, Xiawu Zheng, Jie Chen, Yu Wang, Bin Cui, and Wenming Yang. Protein-ligand interaction prior for binding- aware 3d molecule diffusion models. InThe Twelfth International Conference on Learning Representations, 2024
2024
-
[21]
Benchmarking the reliability of qikprop
Leukothea Ioakimidis, Loizos Thoukydidis, Amin Mirza, Saira Naeem, and Jóhannes Reynisson. Benchmarking the reliability of qikprop. correlation between experimental and predicted values. QSAR & Combinatorial Science, 27(4):445–456, 2008
2008
-
[22]
Structure-based drug design with geometric deep learning.Current Opinion in Structural Biology, 79:102548, 2023
Clemens Isert, Kenneth Atz, and Gisbert Schneider. Structure-based drug design with geometric deep learning.Current Opinion in Structural Biology, 79:102548, 2023
2023
-
[23]
A graph-based genetic algorithm and generative model/monte carlo tree search for the exploration of chemical space.Chemical science, 10(12):3567–3572, 2019
Jan H Jensen. A graph-based genetic algorithm and generative model/monte carlo tree search for the exploration of chemical space.Chemical science, 10(12):3567–3572, 2019
2019
-
[24]
Guided multi-objective generative ai to enhance structure-based drug design.Chemical Science, 16(29): 13196–13210, 2025
Amit Kadan, Kevin Ryczko, Erika Lloyd, Adrian Roitberg, and Takeshi Yamazaki. Guided multi-objective generative ai to enhance structure-based drug design.Chemical Science, 16(29): 13196–13210, 2025
2025
-
[25]
Rdkit documentation.Release, 1(1-79):4, 2013
Greg Landrum. Rdkit documentation.Release, 1(1-79):4, 2013
2013
-
[26]
Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.Advanced drug delivery reviews, 64:4–17, 2012
Christopher A Lipinski, Franco Lombardo, Beryl W Dominy, and Paul J Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.Advanced drug delivery reviews, 64:4–17, 2012
2012
-
[27]
A 3d generative model for structure-based drug design.Advances in Neural Information Processing Systems, 34:6229–6239, 2021
Shitong Luo, Jiaqi Guan, Jianzhu Ma, and Jian Peng. A 3d generative model for structure-based drug design.Advances in Neural Information Processing Systems, 34:6229–6239, 2021
2021
-
[28]
Open babel: An open chemical toolbox.Journal of cheminformatics, 3 (1):33, 2011
Noel M O’Boyle, Michael Banck, Craig A James, Chris Morley, Tim Vandermeersch, and Geoffrey R Hutchison. Open babel: An open chemical toolbox.Journal of cheminformatics, 3 (1):33, 2011
2011
-
[29]
Pocket2mol: Efficient molecular sampling based on 3d protein pockets
Xingang Peng, Shitong Luo, Jiaqi Guan, Qi Xie, Jian Peng, and Jianzhu Ma. Pocket2mol: Efficient molecular sampling based on 3d protein pockets. InInternational conference on machine learning, pages 17644–17655. PMLR, 2022
2022
-
[30]
Hypothesis driven drug design: improving quality and effectiveness of the design-make-test-analyse cycle.Drug discovery today, 17(1-2):56–62, 2012
Alleyn T Plowright, Craig Johnstone, Jan Kihlberg, Jonas Pettersson, Graeme Robb, and Richard A Thompson. Hypothesis driven drug design: improving quality and effectiveness of the design-make-test-analyse cycle.Drug discovery today, 17(1-2):56–62, 2012
2012
-
[31]
Piloting structure-based drug design via modality-specific optimal schedule
Keyue Qiu, Yuxuan Song, Zhehuan Fan, Peidong Liu, Zhe Zhang, Mingyue Zheng, Hao Zhou, and Wei-Ying Ma. Piloting structure-based drug design via modality-specific optimal schedule. InInternational Conference on Machine Learning, pages 50619–50644. PMLR, 2025
2025
-
[32]
Empower structure-based molecule optimization with gradient guided bayesian flow networks
Keyue Qiu, Yuxuan Song, Jie Yu, Hongbo Ma, Ziyao Cao, Zhilong Zhang, Yushuai Wu, Mingyue Zheng, Hao Zhou, and Wei-Ying Ma. Empower structure-based molecule optimization with gradient guided bayesian flow networks. InInternational Conference on Machine Learning, pages 50645–50671. PMLR, 2025. 11
2025
-
[33]
Molcraft: Structure-based drug design in continuous parameter space
Yanru Qu, Keyue Qiu, Yuxuan Song, Jingjing Gong, Jiawei Han, Mingyue Zheng, Hao Zhou, and Wei-Ying Ma. Molcraft: Structure-based drug design in continuous parameter space. In International Conference on Machine Learning, pages 41749–41768. PMLR, 2024
2024
-
[34]
Mollm: Multi-objective large language model for molecular design–optimizing with experts
Nian Ran, Yue Wang, and Richard Allmendinger. Mollm: Multi-objective large language model for molecular design–optimizing with experts. 2025
2025
-
[35]
Plip: fully automated protein–ligand interaction profiler.Nucleic acids research, 43 (W1):W443–W447, 2015
Sebastian Salentin, Sven Schreiber, V Joachim Haupt, Melissa F Adasme, and Michael Schroeder. Plip: fully automated protein–ligand interaction profiler.Nucleic acids research, 43 (W1):W443–W447, 2015
2015
-
[36]
Oleg Trott and Arthur J Olson. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.Journal of computational chemistry, 31(2):455–461, 2010
2010
-
[37]
Deep learning approaches for de novo drug design: An overview.Current opinion in structural biology, 72:135–144, 2022
Mingyang Wang, Zhe Wang, Huiyong Sun, Jike Wang, Chao Shen, Gaoqi Weng, Xin Chai, Honglin Li, Dongsheng Cao, and Tingjun Hou. Deep learning approaches for de novo drug design: An overview.Current opinion in structural biology, 72:135–144, 2022
2022
-
[38]
The strategies and politics of successful design, make, test, and analyze (dmta) cycles in lead generation.Lead Generation, pages 487–512, 2016
Steven S Wesolowski and Dean G Brown. The strategies and politics of successful design, make, test, and analyze (dmta) cycles in lead generation.Lead Generation, pages 487–512, 2016
2016
-
[39]
Tamgen: drug design with target-aware molecule generation through a chemical language model.Nature Communications, 15(1):9360, 2024
Kehan Wu, Yingce Xia, Pan Deng, Renhe Liu, Yuan Zhang, Han Guo, Yumeng Cui, Qizhi Pei, Lijun Wu, Shufang Xie, et al. Tamgen: drug design with target-aware molecule generation through a chemical language model.Nature Communications, 15(1):9360, 2024
2024
-
[40]
Learning subpocket prototypes for generalizable structure-based drug design
Zaixi Zhang and Qi Liu. Learning subpocket prototypes for generalizable structure-based drug design. InInternational Conference on Machine Learning, pages 41382–41398. PMLR, 2023
2023
-
[41]
Structure-based drug design with geometric deep learning: A comprehensive survey
Zaixi Zhang, Jiaxian Yan, Yining Huang, Qi Liu, Enhong Chen, Mengdi Wang, and Marinka Zitnik. Structure-based drug design with geometric deep learning: A comprehensive survey. ACM Computing Surveys, 58(5):1–35, 2025
2025
-
[42]
preserve affinity while improving druggability
Jingyuan Zhou, Dengwei Zhao, Hao Qian, Shikui Tu, and Lei Xu. Multi-objective structure- based drug design using causal discovery.IEEE Transactions on Computational Biology and Bioinformatics, 2025. 12 A Prompts Prompt A.1: MOO-awareness prompt injected into the design stage of CIDD. [Multi-Objective Optimization Guidance] You are optimizing a ligand agai...
2025
-
[43]
Identify all optimization goals mentioned in the reasoning
-
[44]
If only affinity-related goals are mentioned, output: affinity
-
[45]
If only druggability-related goals are mentioned, output: druggability
-
[46]
the other is described as a side benefit, a constraint to preserve, or a minor consideration), output the dominant one
If both are mentioned but one is the main driver of the edit (e.g. the other is described as a side benefit, a constraint to preserve, or a minor consideration), output the dominant one
-
[47]
Only output "both" when the reasoning treats affinity and druggability as co-equal targets of the same edit
-
[48]
general background, descriptions of the pocket, summaries of prior rounds) when deciding the intent
Ignore meta-commentary that is not about this specific edit (e.g. general background, descriptions of the pocket, summaries of prior rounds) when deciding the intent. Reasoning text: {reasoning} Prompt A.3: Prompt for PROBE in site map construction. You are an elite Director of Medicinal Chemistry, Expert Toxicologist, and Structural Biologist. Your task ...
-
[49]
Fused ring systems should not be completely saturated (all sp3 carbons)
-
[50]
Ring carbon atoms should ideally have consistent hybridization
-
[51]
[Input Data]
Reactive groups (e.g., Michael acceptors, anilines) count as structural alerts. [Input Data]
-
[52]
Structure SMILES : {smi}
-
[53]
Mapped SMILES : {mapped_smi}
-
[54]
Binding Metrics : Vina Score {score} | Ligand Efficiency (LE) {mc_data['le']:.3f}
-
[55]
Properties : MW {mc_data['mw']:.1f} | LogP {mc_data['logp']:.2f} | TPSA {mc_data['tpsa']:.1f}
-
[56]
Complexity : Chiral Centers {mc_data['chiral_centers']} | BertzCT {mc_data['sa_proxy']:.1f}
-
[57]
Flatten this complex sp3 ring system
Structural Alerts: {','.join(mc_data['alerts']) if mc_data['alerts'] else 'Clean'} 14 [Diagnosis Signals] A. Interaction signals (PLIP): {interaction_text} B. Geometric signals -- Clashes: {clash_text} C. Geometric signals -- Voids / Buriedness: {void_text} D. Geometric signals -- Chemical Mismatch: {mismatch_text} E. Fragment-level interaction mapping (B...
-
[58]
Base each strategy strictly on the chemical nature of the sites in the Site Map
NO PROMPT COPYING. Base each strategy strictly on the chemical nature of the sites in the Site Map
-
[59]
Each strategy must be expressed as a chemically explicit edit prescription (e.g., extending an H-bond donor, hopping to an achiral bioisostere, ring-opening, pruning)
ABSTRACT OPERATIONS ONLY. Each strategy must be expressed as a chemically explicit edit prescription (e.g., extending an H-bond donor, hopping to an achiral bioisostere, ring-opening, pruning)
-
[60]
"" target_strategies = ['A','B','C'] for stg_key in target_strategies: stg_content = strategy_dict[stg_key] if stg_key =='A': tactic_instructions = r
STRUCTURAL-ALERT / COMPLEXITY COMPLIANCE. Unless the strategy explicitly accepts druggability damage (Strategy A), structural modifications should reduce complexity, avoid unnecessary chiral centers, and avoid spiro / bridged / fully-sp3 fused ring systems. [Task: Emit three Strategies sigma_k = < V_k, pi_k, tau_k >] For each strategy, output exactly the ...
-
[61]
High intensity
"High intensity" -- Fill pocket void with ring. Substantial volume addition (Delta N_heavy >= +4); insert a complete standard ring system (e.g., phenyl / pyridyl / morpholino) into a void identified for sigma_A
-
[62]
Medium intensity
"Medium intensity" -- Add functional group. Moderate volume addition (+1 <= Delta N_heavy <= +3); attach a small group (isopropyl, CF3, amide linker)
-
[63]
Low intensity
"Low intensity" -- Isosteric tweak. 17 Minimal volume change (Delta N_heavy in {0, +1}); bioisosteric substitution (-CH3 -> -CF3, phenyl -> pyridyl) to retune electrostatics
-
[64]
Counterfactual
"Counterfactual" -- Delete functional group. Reverse-direction probe (Delta N_heavy <= -1): remove a key H-bond donor / acceptor or anchor. Expected signal: if Vina worsens, the original group is load-bearing; if Vina is unchanged, the strategy probes a non-causal direction; if Vina improves, the targeted group is actively harmful. """ elif stg_key =='B':...
-
[65]
High intensity
"High intensity" -- Prune Liability / Tension sites. Aggressive pruning (Delta N_heavy <= -4); remove bulky lipophilic groups, open complex fused rings into a single ring, or strip multiple chiral centers in one move
-
[66]
Medium intensity
"Medium intensity" -- Trim peripheral liabilities. Surgical pruning (-3 <= Delta N_heavy <= -1); drop redundant terminals or simplify a substituted ring into an unsubstituted standard ring
-
[67]
Low intensity
"Low intensity" -- Shave solvent-exposed atoms. Minimal pruning (Delta N_heavy = -1); remove a single solvent-exposed atom (terminal methyl / halogen) that contributes no binding energy
-
[68]
Counterfactual
"Counterfactual" -- Add sp3 bloat. Reverse-direction probe (Delta N_heavy >= +3): attach a synthetically difficult, unnecessary sp3-rich bulky group (tert-butyl / cyclopentyl) to a solvent-exposed atom. Expected signal: if SA / QED collapse without Vina gain, mass at this site is purely harmful, confirming the pruning direction is causal. """ else: # stg_...
-
[69]
High intensity
"High intensity" -- Replace core scaffold. Major topological shift with bounded mass change (-2 <= Delta N_heavy <= +2); swap a central ring or core linker for a 18 fundamentally different standard ring (phenyl -> tetrahydropyran / pyrimidine / piperazine) to reshape 3D geometry and Fsp3 without molecular-weight bloat
-
[70]
Medium intensity
"Medium intensity" -- Peripheral bioisostere swap. Local topological shift (-1 <= Delta N_heavy <= +1); replace a peripheral group with a recognized bioisostere (carboxylic acid -> tetrazole, amide -> 1,2,4-triazole)
-
[71]
Low intensity
"Low intensity" -- Regio-isomeric shift. No mass change (Delta N_heavy = 0); keep the groups, alter connectivity (ortho -> meta / para; reverse an amide -C(=O)NH- -> -NHC(=O)-)
-
[72]
Counterfactual
"Counterfactual" -- Break geometric constraint. Deliberately violate a mandatory geometry: convert a planar aromatic ring essential for pi-stacking into a saturated ring (benzene -> cyclohexane), or rigidify a flexible linker via an alkyne to disrupt induced fit. Expected signal: if Vina collapses, the original rigid / planar topology is load-bearing for ...
-
[73]
Target: [one atom index]
"Grow": Attach a new fragment by replacing an implicit hydrogen on the target. Target: [one atom index]
-
[74]
Replace_Terminal_Group
"Replace_Terminal_Group": Cut a peripheral group (-OH, -CH3, halogen, ...) and swap it. Target: [one atom index of the group to be removed]
-
[75]
Replace_Sidechain_or_Ring
"Replace_Sidechain_or_Ring": 19 Cut an entire sidechain or ring system attached to the main scaffold. Target: [one atom index -- MUST be the anchoring atom of the sidechain / ring]
-
[76]
Delete_Group
"Delete_Group": Remove a peripheral group without replacement (bond capped by H). Target: [one atom index]. [Fragment Constraints -- SEMANTIC TAGS ONLY] Do NOT output raw numbers for physicochemical properties. Use these tags: - size : "Small" (MW < 150) / "Medium" (150-250) / "Large" (> 250) / "Any" - polarity : "Hydrophilic" / "Neutral" / "Lipophilic" /...
-
[77]
Index every entry by the abstract site (s_1..s_n), not by atom indices and not by strategy name
SITE-LEVEL ABSTRACTION. Index every entry by the abstract site (s_1..s_n), not by atom indices and not by strategy name. The molecule mutates across rounds; atom indices are not stable, but each site is named after its originating fragment / pharmacophore role
-
[78]
For each site, pull together every probe whose target_site equals this site -- possibly drawn from multiple strategies
EVIDENCE AGGREGATION. For each site, pull together every probe whose target_site equals this site -- possibly drawn from multiple strategies. Cite probe_ids when stating a verdict
-
[79]
For each strategy sigma_k, state whether the observed forward_shape / counterfactual_signal validates or invalidates tau_k (the trade-off sigma_k claimed to accept)
STRATEGY VERDICT. For each strategy sigma_k, state whether the observed forward_shape / counterfactual_signal validates or invalidates tau_k (the trade-off sigma_k claimed to accept)
-
[80]
Translate physical responses into the 5 semantic tags used by the Fragment Assembly Engine: size / polarity / flexibility / shape / charge
SEMANTIC ENVELOPES. Translate physical responses into the 5 semantic tags used by the Fragment Assembly Engine: size / polarity / flexibility / shape / charge
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.