Benchmark Dataset for Catalysis on 2D MXenes
Pith reviewed 2026-06-28 18:10 UTC · model grok-4.3
The pith
Machine learning interatomic potentials trained on 60,000 DFT calculations predict MXene forces and energies at meV accuracy while delivering 1000- to 4000-fold CPU speedup.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
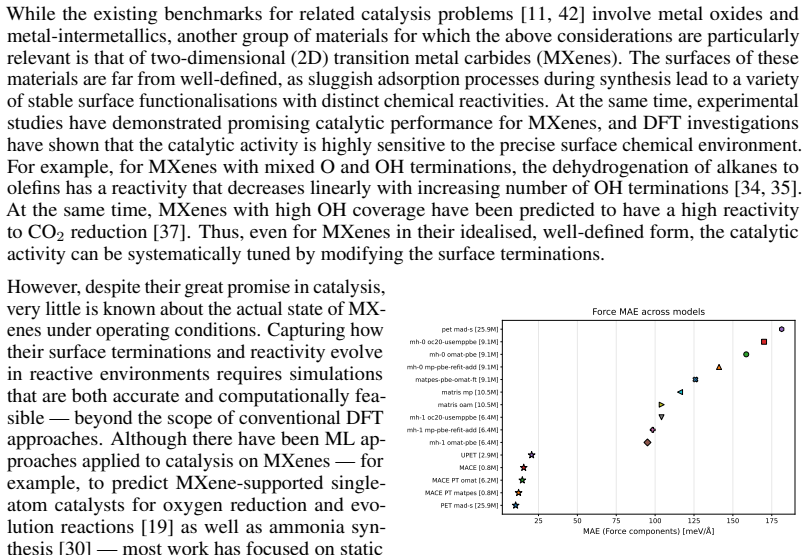
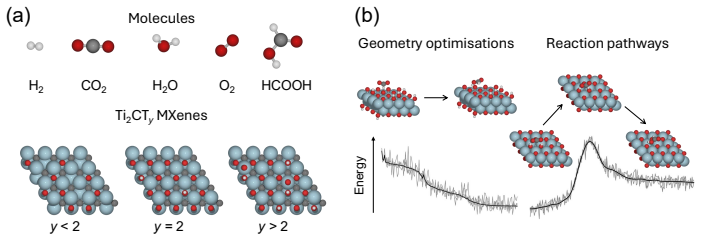
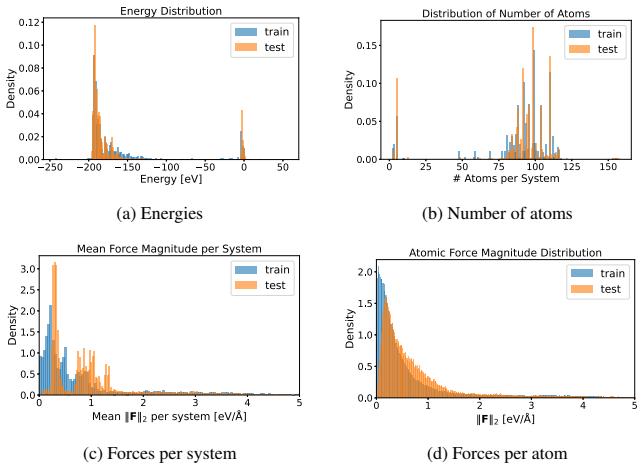
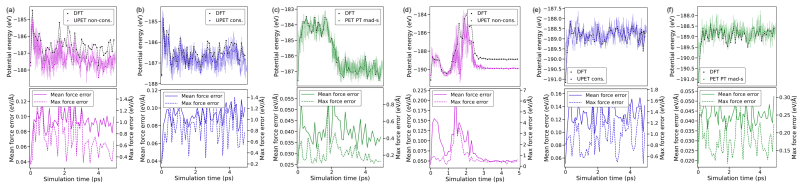
A combined DFT-ML framework built on 50,000 training and 10,000 test DFT calculations for Ti2CTy MXenes and molecules, together with 1,000 new larger systems, allows EquiformerV2, MACE, MatRIS, and UPET models to replace repeated first-principles force and energy evaluations. The models maintain approximately 10 meV/Å force accuracy and 1 meV per-atom energy accuracy while providing 1-4·10^3 computational acceleration on CPU, thereby enabling more extensive catalytic simulations of 2D MXenes than direct DFT permits.
What carries the argument
Machine learning interatomic potentials trained on a large DFT dataset of MXene and molecular configurations to replace repeated first-principles force and energy calculations.
If this is right
- Structural relaxations and molecular-dynamics trajectories of MXene surfaces become feasible at scales previously limited by DFT cost.
- High-throughput screening of different surface terminations and adsorbates on MXenes can be performed with the reported speedup.
- The released dataset serves as a public benchmark for future MLIP development targeted at 2D catalytic materials.
- Qualitative simulation-based checks become a necessary complement to numerical error metrics when validating models for catalysis.
- The 1,000-system generalization test provides a concrete protocol for assessing transferability to larger MXene models.
Where Pith is reading between the lines
- The same dataset-plus-MLIP strategy could be repeated for other families of 2D materials once equivalent DFT reference data exist.
- Coupling the fast ML potentials to Monte Carlo or grand-canonical sampling would allow direct prediction of surface coverage under realistic gas pressures.
- Active-learning loops seeded by the existing 60,000-point set could further reduce the number of new DFT calculations needed for specific reaction pathways.
- The reported CPU speedup makes it practical to embed the potentials inside larger multiscale models that link atomic catalysis to mesoscale transport.
Load-bearing premise
The trained models will retain the target accuracy when applied to genuinely new and larger MXene systems outside the training distribution.
What would settle it
If force or energy predictions on the 1,000 new larger systems exceed roughly 10 meV/Å or 1 meV per atom relative to fresh DFT reference values, the maintained-accuracy claim is falsified.
Figures
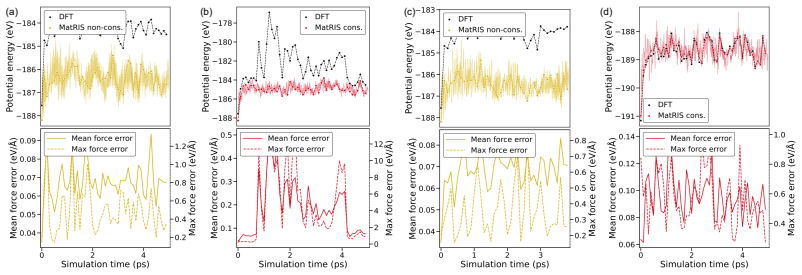
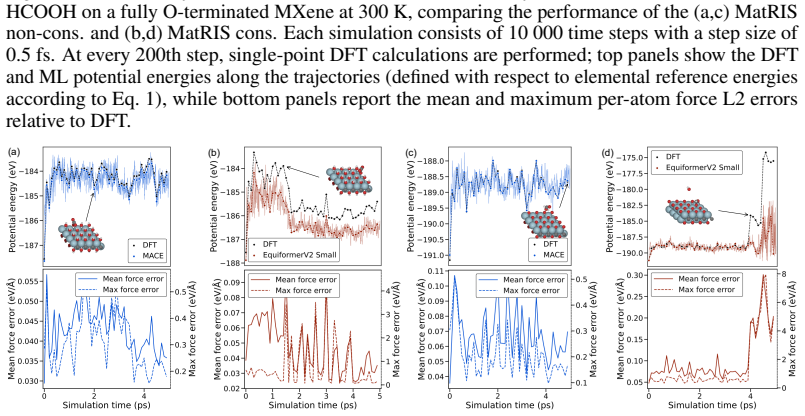
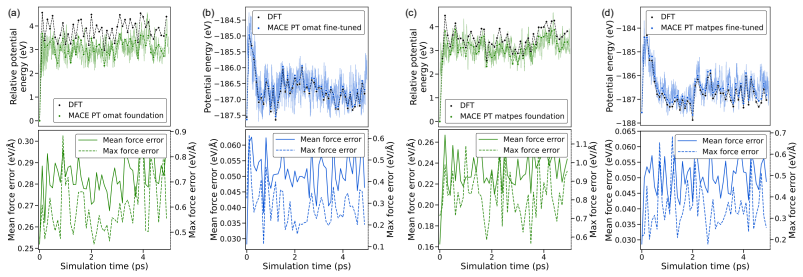
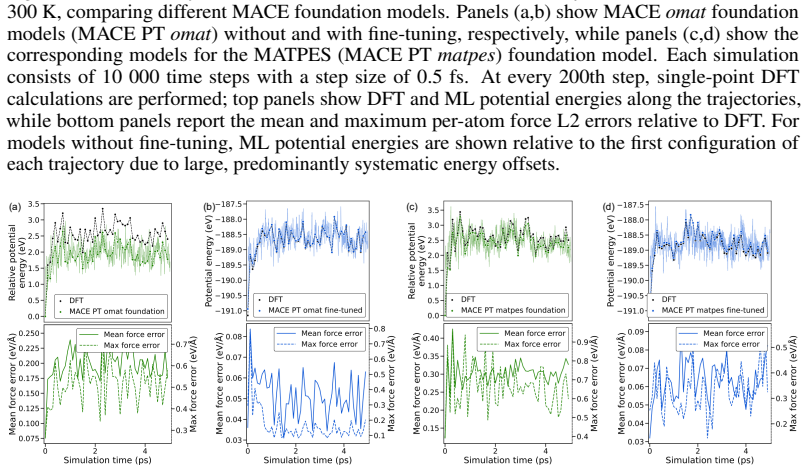
read the original abstract
Merging first-principles calculations with machine learning (ML), we aim to accelerate the exploration of catalytic behaviour in novel materials. We focus on two-dimensional (2D) Ti$_2$CT$_y$ MXenes, whose versatile surface chemistry makes them particularly compelling candidates for catalysis. Resolving their composition and structure under realistic conditions exceeds the reach of standard density functional theory (DFT) due to computational cost. To address this challenge, we generate a comprehensive dataset of 50,000 DFT calculations for training and 10,000 for testing, encompassing both Ti$_2$CT$_y$ MXene configurations and molecular systems, along with an additional test dataset with 1000 genuinely new, larger systems to investigate how well models generalise. We train and validate widely used and competitive machine learning interatomic potential (MLIP) models, including EquiformerV2, MACE, MatRIS, and UPET, that accurately predict atomic forces and formation energies -- quantities that DFT must repeatedly compute for structural and catalytic investigations -- for these 2D materials. This combined DFT-ML framework achieves computational acceleration on the order of approximately $1-4 \cdot 10^3$ (on a CPU) while maintaining desired-level accuracy (approximately +/- $10$ meV/A for forces and approximately +/- $1$ meV for per-atom energies), paving the way for more efficient investigations of MXene catalytic behaviour. Moreover, we perform an extensive qualitative evaluation of the trained models, showcasing the importance of comprehensive simulation-based comparison beyond benchmark metrics. The dataset and the trained models with the code are available at https://huggingface.co/datasets/CatalystAnonymous/catalyst_mxenes.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a benchmark dataset comprising 50,000 DFT calculations for training and 10,000 for testing on Ti₂CTy MXene configurations and molecular systems relevant to catalysis, plus a separate 1,000-system hold-out of genuinely larger structures. Several ML interatomic potentials (EquiformerV2, MACE, MatRIS, UPET) are trained to predict forces and formation energies, with the combined DFT-ML approach claimed to deliver 1–4 × 10³ CPU speedup while retaining target accuracy of approximately ±10 meV/Å for forces and ±1 meV/atom for energies. The dataset, trained models, and code are released publicly.
Significance. If the reported accuracy generalizes, the work supplies a substantial, openly available resource that could materially accelerate high-throughput screening of MXene catalytic properties, where repeated DFT evaluations of forces and energies are the dominant cost. The public release of both the 60k+ DFT dataset and the trained MLIP checkpoints constitutes a concrete, reusable contribution beyond the benchmark numbers themselves.
major comments (2)
- [Abstract] Abstract: The 1,000 genuinely new, larger systems are explicitly introduced “to investigate how well models generalise,” yet the manuscript supplies no quantitative error metrics (MAE, RMSE, or force/energy histograms) on this hold-out set. Because catalytic investigations routinely involve structures larger than the 50k/10k training distribution, the absence of these numbers leaves the central claim that the framework “maintains desired-level accuracy” on realistic systems unverified.
- [Abstract] Abstract and methods: The stated accuracy targets (±10 meV/Å forces, ±1 meV/atom energies) and the 1–4 × 10³ speedup are presented without accompanying details on DFT convergence criteria, k-point sampling, or error-bar methodology for the reference calculations. Without these controls it is impossible to judge whether the ML models are being benchmarked against sufficiently converged targets or whether the reported errors already incorporate DFT uncertainty.
minor comments (2)
- [Abstract] Abstract: The force accuracy is written as “+/- 10 meV/A”; the conventional unit is meV/Å.
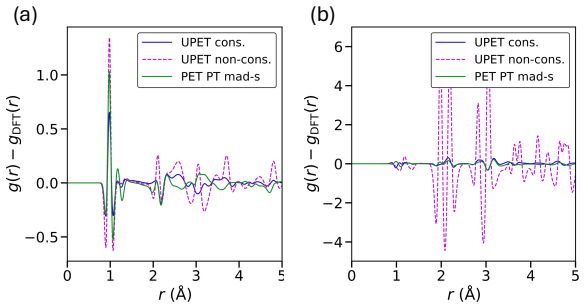
- The manuscript states that an “extensive qualitative evaluation” of the models is performed, but the corresponding figures or supplementary material are not referenced in the provided text.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address each major comment below and will incorporate the requested information into the revised version.
read point-by-point responses
-
Referee: [Abstract] Abstract: The 1,000 genuinely new, larger systems are explicitly introduced “to investigate how well models generalise,” yet the manuscript supplies no quantitative error metrics (MAE, RMSE, or force/energy histograms) on this hold-out set. Because catalytic investigations routinely involve structures larger than the 50k/10k training distribution, the absence of these numbers leaves the central claim that the framework “maintains desired-level accuracy” on realistic systems unverified.
Authors: We agree that quantitative metrics on the 1,000 larger hold-out systems are necessary to support the generalization claims. The revised manuscript will add MAE, RMSE, and force/energy error histograms specifically for this hold-out set, enabling direct evaluation of model performance on structures outside the original training distribution. revision: yes
-
Referee: [Abstract] Abstract and methods: The stated accuracy targets (±10 meV/Å forces, ±1 meV/atom energies) and the 1–4 × 10³ speedup are presented without accompanying details on DFT convergence criteria, k-point sampling, or error-bar methodology for the reference calculations. Without these controls it is impossible to judge whether the ML models are being benchmarked against sufficiently converged targets or whether the reported errors already incorporate DFT uncertainty.
Authors: We acknowledge this omission. The revised manuscript will include a new subsection in the Methods detailing the DFT convergence criteria (energy and force thresholds), k-point sampling settings, and any error estimation procedures used for the reference calculations. This will confirm that the reported ML accuracies are measured against sufficiently converged DFT targets. revision: yes
Circularity Check
No circularity; accuracy and speedup are direct held-out comparisons.
full rationale
The paper generates a 50k/10k DFT dataset split, trains standard MLIPs (EquiformerV2, MACE, etc.), and reports force/energy errors as direct MAE/RMSE against the held-out 10k DFT labels. Speedup (1-4·10³ on CPU) is a timing ratio between DFT and ML inference. The 1000 larger systems are introduced only to probe generalization and carry no reported metrics that feed back into the accuracy claim. No equations, fitted parameters, or self-citations reduce any reported number to a tautology or to the training inputs by construction. The derivation chain is therefore self-contained against external DFT benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption DFT calculations with the chosen functional and settings provide sufficiently accurate reference forces and energies for training MLIPs intended for catalysis studies.
Reference graph
Works this paper leans on
-
[1]
Anderson, T
B. Anderson, T. S. Hy, and R. Kondor. Cormorant: Covariant molecular neural networks. Advances in Neural Information Processing Systems, pages 14510–14519, 2019
2019
-
[2]
Aykent and T
S. Aykent and T. Xia. Gotennet: Rethinking efficient 3d equivariant graph neural networks.The Thirteenth International Conference on Learning Representations, 2025
2025
-
[3]
Barroso-Luque, S
L. Barroso-Luque, S. Muhammed, X. Fu, B. M. Wood, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick, and Z. W. Ulissi. Open materials 2024 (omat24) inorganic materials dataset and models. arXiv, 2024
2024
-
[4]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Cs´anyi. MACE: Higher order equivari- ant message passing neural networks for fast and accurate force fields.Advances in neural information processing systems, 35:11423–11436, 2022
2022
-
[5]
Batatia, P
I. Batatia, P. Benner, Y . Chiang, A. M. Elena, D. P. Kov ´acs, J. Riebesell, X. R. Advincula, M. Asta, W. J. Baldwin, N. Bernstein, A. Bhowmik, S. M. Blau, V . C˘arare, J. P. Darby, S. De, F. D. Pia, V . L. Deringer, R. Elijoˇsius, Z. El-Machachi, E. Fako, A. C. Ferrari, A. Genreith- Schriever, J. George, R. E. A. Goodall, C. P. Grey, S. Han, W. Handley,...
2023
-
[6]
Batzner, A
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nat. Commun., 13(1):2453, 2022
2022
-
[7]
F. Bigi, P. Pegolo, A. Mazitov, and M. Ceriotti. Pushing the limits of unconstrained machine- learned interatomic potentials, 2026. URLhttps://arxiv.org/abs/2601.16195
Pith/arXiv arXiv 2026
-
[8]
P. E. Bl¨ochl. Projector augmented-wave method.Phys. Rev. B, 50(24):17953–17979, Dec. 1994. ISSN 0163-1829, 1095-3795. doi: 10.1103/PhysRevB.50.17953. URL https://link.aps. org/doi/10.1103/PhysRevB.50.17953
-
[9]
M. M. Bronstein, J. Bruna, Y . LeCun, A. Szlam, and P. Vandergheynst. Geometric deep learning: going beyond euclidean data.IEEE Signal Processing Magazine, 34(4):18–42, 2017
2017
-
[10]
M. M. Bronstein, J. Bruna, T. Cohen, and P. Veliˇckovi´c. Geometric deep learning: Grids, groups, graphs, geodesics, and gauges.arXiv preprint arXiv:2104.13478, 2021. 10
Pith/arXiv arXiv 2021
-
[11]
L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick, and Z. Ulissi. Open catalyst 2020 (OC20) dataset and community challenges.ACS Catal., 11(10): 6059–6072, May 2021. ISSN 2155-5435, 2155-5435. doi: 10.1021/acscatal.0c04525. URL htt...
-
[12]
Chen and S
C. Chen and S. P. Ong. A universal graph deep learning interatomic potential for the periodic table.Nat. Comput. Sci., 2(11):718–728, 2022
2022
-
[13]
L. Chen, J. Rosen, and J. Bj ¨ork. A density functional benchmark for dehydrogenation and dehalogenation reactions on coinage metal surfaces.ChemPhysChem, 26(1):e202400865, Jan. 2025. ISSN 1439-4235, 1439-7641. doi: 10.1002/cphc.202400865. URL https:// chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cphc.202400865
-
[14]
V . L. Deringer, M. A. Caro, and G. Cs´anyi. Machine learning interatomic potentials as emerging tools for materials science.Adv. Mater ., 31(46):1902765, 2019
2019
-
[15]
M. Dion, H. Rydberg, E. Schr¨oder, D. C. Langreth, and B. I. Lundqvist. Van der waals density functional for general geometries.Phys. Rev. Lett., 92(24):246401, June 2004. ISSN 0031-9007, 1079-7114. doi: 10.1103/PhysRevLett.92.246401. URL https://link.aps.org/doi/10. 1103/PhysRevLett.92.246401
-
[16]
Esteves, C
C. Esteves, C. Allen-Blanchette, A. Makadia, and K. Daniilidis. Learning SO(3) Equivariant Representations With Spherical CNNs.CoRR, 2017. URL http://arxiv.org/abs/1711. 06721
2017
-
[17]
X. Fu, B. M. Wood, L. Barroso-Luque, D. S. Levine, M. Gao, M. Dzamba, and C. L. Zitnick. Learning smooth and expressive interatomic potentials for physical property prediction. In International Conference on Machine Learning, pages 17875–17893. PMLR, 2025
2025
-
[18]
Fuchs, D
F. Fuchs, D. Worrall, V . Fischer, and M. Welling. Se(3)-transformers: 3d roto-translation equivariant attention networks.Advances in Neural Information Processing Systems, 33:1970– 1981, 2020
1970
-
[19]
H. Guo and S. G. Lee. Machine learning-guided discovery of thermodynamically stable single-atom catalysts on functionalized MXenes for enhanced oxygen reduction and evolution reactions.J. Mater . Chem. A, 13(28):22730–22744, 2025. ISSN 2050-7488, 2050-7496. doi: 10.1039/D5TA02929E. URLhttps://xlink.rsc.org/?DOI=D5TA02929E
-
[20]
I. Hamada. van der waals density functional made accurate.Phys. Rev. B, 89(12):121103,
-
[21]
doi: 10.1103/PhysRevB.89.121103
ISSN 1098-0121, 1550-235X. doi: 10.1103/PhysRevB.89.121103. URL https: //link.aps.org/doi/10.1103/PhysRevB.89.121103
-
[22]
Hohenberg and W
P. Hohenberg and W. Kohn. Inhomogeneous electron gas.Phys. Rev., 136(3B):B864–B871,
-
[23]
ISSN 0031-899X. doi: 10.1103/PhysRev.136.B864. URL https://link.aps.org/ doi/10.1103/PhysRev.136.B864
-
[24]
A. D. Kaplan, R. Liu, J. Qi, T. W. Ko, B. Deng, J. Riebesell, G. Ceder, K. A. Persson, and S. P. Ong. A foundational potential energy surface dataset for materials, 2025
2025
-
[25]
Self-consistent equations including exchange and correlation effects,
W. Kohn and L. J. Sham. Self-consistent equations including exchange and correlation effects. Phys. Rev., 140(4A):A1133–A1138, 1965. ISSN 0031-899X. doi: 10.1103/PhysRev.140.A1133. URLhttps://link.aps.org/doi/10.1103/PhysRev.140.A1133
-
[26]
G. Kresse and J. Furthm¨uller. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set.Phys. Rev. B, 54(16):11169–11186, Oct. 1996. ISSN 0163- 1829, 1095-3795. doi: 10.1103/PhysRevB.54.11169. URL https://link.aps.org/doi/ 10.1103/PhysRevB.54.11169
-
[27]
G. Kresse and D. Joubert. From ultrasoft pseudopotentials to the projector augmented-wave method.Phys. Rev. B, 59(3):1758–1775, Jan. 1999. ISSN 0163-1829, 1095-3795. doi: 10.1103/ PhysRevB.59.1758. URLhttps://link.aps.org/doi/10.1103/PhysRevB.59.1758
-
[28]
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Sch ¨u...
2017
-
[29]
D. S. Levine, M. Shuaibi, E. W. C. Spotte-Smith, M. G. Taylor, M. R. Hasyim, K. Michel, I. Batatia, G. Cs ´anyi, M. Dzamba, P. Eastman, N. C. Frey, X. Fu, V . Gharakhanyan, A. S. Krishnapriyan, J. A. Rackers, S. Raja, A. Rizvi, A. S. Rosen, Z. Ulissi, S. Vargas, C. L. Zitnick, S. M. Blau, and B. M. Wood. The open molecules 2025 (omol25) dataset, evaluatio...
arXiv 2025
-
[30]
Liao and T
Y .-L. Liao and T. Smidt. Equiformer: Equivariant Graph Attention Transformer for 3D Atomistic Graphs.The Eleventh International Conference on Learning Representations, 2023
2023
-
[31]
Y .-L. Liao, B. Wood, A. Das, and T. Smidt. EquiformerV2: Improved Equivariant Trans- former for Scaling to Higher-Degree Representations.The Twelfth International Conference on Learning Representations, 2024
2024
-
[32]
G. Lin, T. Guo, W. Lin, H. Fan, L. Guo, Z. Zhang, B. Li, J. Wang, H. Ji, W. Song, and J. Fu. Machine learning accelerated screening advanced single-atom anchored MXenes electrocat- alyst for nitrogen fixation.ACS Catal., 15(15):13534–13548, Aug. 2025. ISSN 2155-5435, 2155-5435. doi: 10.1021/acscatal.4c06914. URL https://pubs.acs.org/doi/10.1021/ acscatal.4c06914
-
[33]
C. Malosso, F. Bigi, P. Pegolo, J. W. Abbott, P. Loche, M. Rossi, M. Ceriotti, and A. Mazitov. High-quality, high-information datasets for universal atomistic machine learning, 2026. URL https://arxiv.org/abs/2603.02089
arXiv 2026
-
[34]
Melnyk, M
P. Melnyk, M. Felsberg, M. Wadenb¨ack, A. Robinson, and C. Le. O n Learning Deep O(n)- Equivariant Hyperspheres.Proceedings of the 41st International Conference on Machine Learning, 235:35324–35339, 7 2024. URL https://proceedings.mlr.press/v235/ melnyk24a.html
2024
-
[35]
Melnyk, A
P. Melnyk, A. Robinson, M. Felsberg, and M. Wadenb¨ack. TetraSphere: A Neural Descriptor for O(3)-Invariant Point Cloud Analysis.Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), pages 5620–5630, 6 2024
2024
-
[36]
K. Niu, L. Chi, J. Rosen, and J. Bj¨ork. C–h activation of light alkanes on mxenes predicted by hy- drogen affinity.Phys. Chem. Chem. Phys., 22(33):18622–18630, 2020. ISSN 1463-9076, 1463-
2020
-
[37]
URLhttps://xlink.rsc.org/?DOI=D0CP02471F
doi: 10.1039/D0CP02471F. URLhttps://xlink.rsc.org/?DOI=D0CP02471F
-
[38]
K. Niu, L. Chi, J. Rosen, and J. Bj ¨ork. Structure-activity correlation of Ti 2CT2 mxenes for c–h activation.J. Phys. Condens. Matter, 33(23):235201, June 2021. ISSN 0953-8984, 1361- 648X. doi: 10.1088/1361-648X/abe8a1. URL https://iopscience.iop.org/article/ 10.1088/1361-648X/abe8a1
-
[39]
K. Niu, J. Bj ¨ork, and J. Rosen. First-principles exploration of Sc- and Y-based MX- enes with halogen terminations.npj 2D Mater . Appl., 9(1):69, 2025. ISSN 2397-
2025
-
[40]
URL https://www.nature.com/articles/ s41699-025-00589-7
doi: 10.1038/s41699-025-00589-7. URL https://www.nature.com/articles/ s41699-025-00589-7
-
[41]
A. Parui, P. Srivastava, and A. K. Singh. Selective reduction of CO 2 on Ti2C(OH)2 MXene through spontaneous crossing of transition states.ACS Appl. Mater . Interfaces, 14(36):40913– 40920, 2022. ISSN 1944-8244, 1944-8252. doi: 10.1021/acsami.2c10213. URL https: //pubs.acs.org/doi/10.1021/acsami.2c10213
-
[42]
Passaro and C
S. Passaro and C. L. Zitnick. Reducing SO(3) convolutions to SO(2) for efficient equivariant GNNs.Proceedings of the 40th International Conference on Machine Learning, 202:27420– 27438, 7 2023. URLhttps://proceedings.mlr.press/v202/passaro23a.html
2023
-
[43]
Paszke, S
A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, et al. Pytorch: An imperative style, high-performance deep learning library.Advances in Neural Information Processing Systems, pages 8024–8035, 2019
2019
-
[44]
D. Ruhe, J. Brandstetter, and P. Forr ´e. Clifford Group Equivariant Neural Networks. Thirty-seventh Conference on Neural Information Processing Systems, 2023. URL https: //openreview.net/forum?id=n84bzMrGUD
2023
-
[45]
F. Tran, L. Kalantari, B. Traor ´e, X. Rocquefelte, and P. Blaha. Nonlocal van der Waals functionals for solids: Choosing an appropriate one.Phys. Rev. Materials, 3(6):063602, June
-
[46]
doi: 10.1103/PhysRevMaterials.3.063602
ISSN 2475-9953. doi: 10.1103/PhysRevMaterials.3.063602. URL https://link.aps. org/doi/10.1103/PhysRevMaterials.3.063602. 12
-
[47]
R. Tran, J. Lan, M. Shuaibi, B. M. Wood, S. Goyal, A. Das, J. Heras-Domingo, A. Kolluru, A. Rizvi, N. Shoghi, A. Sriram, F. Therrien, J. Abed, O. V oznyy, E. H. Sargent, Z. Ulissi, and C. L. Zitnick. The open catalyst 2022 (oc22) dataset and challenges for oxide electrocatalysts. ACS Catal., 13(5):3066–3084, 2023. doi: 10.1021/acscatal.2c05426. URL https:...
-
[48]
Van Gool, T
L. Van Gool, T. Moons, E. Pauwels, and A. Oosterlinck. Vision and lie’s approach to invariance. Image and vision computing, 13(4):259–277, 1995
1995
-
[49]
G. Wang, C. Wang, X. Zhang, Z. Li, J. Zhou, and Z. Sun. Machine learning interatomic potential: Bridge the gap between small-scale models and realistic device-scale simulations. Iscience, 27(5), 2024
2024
-
[50]
Weiler, M
M. Weiler, M. Geiger, M. Welling, W. Boomsma, and T. S. Cohen. 3D steerable CNNs: Learning rotationally equivariant features in volumetric data.Advances in Neural Information Processing Systems, pages 10381–10392, 2018
2018
-
[51]
Weiler, P
M. Weiler, P. Forr´e, E. Verlinde, and M. Welling. Equivariant and coordinate independent convolutional networks.A Gauge Field Theory of Neural Networks, page 110, 2023
2023
-
[52]
H. Yang, C. Hu, Y . Zhou, X. Liu, Y . Shi, J. Li, G. Li, Z. Chen, S. Chen, C. Zeni, et al. MatterSim: A deep learning atomistic model across elements, temperatures and pressures.arXiv preprint arXiv:2405.04967, 2024
Pith/arXiv arXiv 2024
-
[53]
Y . Zhou, S. Hu, X. Zhang, H. Wang, G. Tan, and W. Jia. MatRIS: Toward reliable and efficient pretrained machine learning interatomic potentials. InThe F ourteenth International Conference on Learning Representations, 2026. URL https://openreview.net/forum? id=5xBT5Ziute. 13 A Reference Energy in DFT Dilemma In principle, density functional theory (DFT) p...
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.