Deciphering borophene growth pathways with data-driven simulations
Pith reviewed 2026-06-28 08:58 UTC · model grok-4.3
The pith
Machine-learned potentials in Monte Carlo simulations map kinetic pathways that select specific borophene phases on silver.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
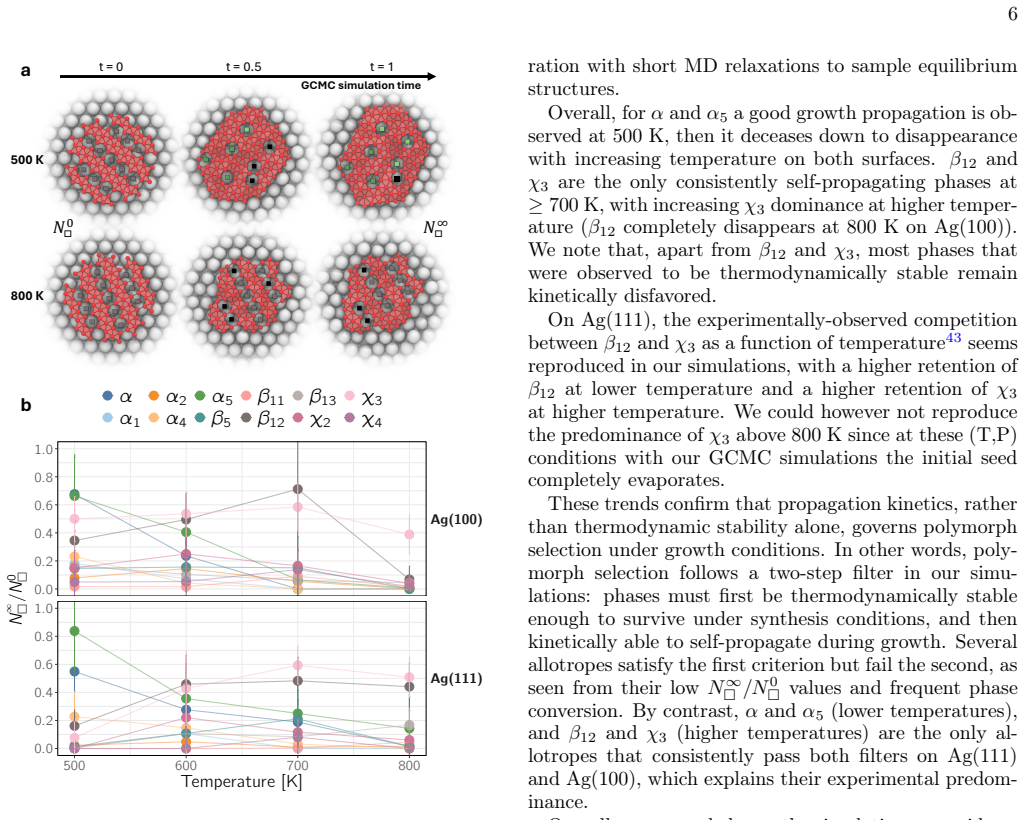
Coupling a reactive machine-learned interatomic potential with grand-canonical Monte Carlo simulations and data-driven structural classification tracks borophene formation on Ag(111) and Ag(100) from early nuclei to extended layers, builds temperature-pressure growth maps, resolves the roles of vacancy motifs, phase intermixing and seed structure in polymorph selection, reproduces the prevalence of β12/χ3 phases and their temperature-dependent competition, and identifies conditions that suppress competing motifs while promoting targeted phases.
What carries the argument
Reactive machine-learned interatomic potential deployed inside grand-canonical Monte Carlo simulations, paired with data-driven structural classification to label phases during growth.
If this is right
- Temperature can be used to tip the balance between β12 and χ3 phases during growth.
- Seed structure and vacancy motifs steer which polymorph reaches long-range order.
- Specific temperature-pressure regions on each silver face suppress unwanted motifs.
- Actionable synthesis windows emerge that favor one targeted phase over its competitors.
Where Pith is reading between the lines
- The same simulation-plus-classification loop could be applied to other 2D materials whose polymorphs compete during nucleation.
- If the predicted windows are confirmed by growth experiments, the approach supplies a general route to reduce polymorphism in low-dimensional systems.
- The identified role of metastable nuclei suggests that pre-seeding substrates with particular motifs could further narrow phase selection.
Load-bearing premise
The machine-learned potential accurately reproduces the energies, barriers, and silver-substrate interactions of boron atoms across the temperatures and pressures of actual growth experiments.
What would settle it
An experiment performed inside one of the predicted temperature-pressure windows on Ag(111) that produces a dominant phase different from the one the simulation forecasts for those conditions.
Figures
read the original abstract
Deterministic synthesis of borophene remains challenging because many polymorphs compete during nucleation and growth. Here we combine a reactive machine-learned interatomic potential with grand-canonical Monte Carlo simulations and data-driven structural classification to track borophene formation from early nuclei to extended layers on Ag(111) and Ag(100). We build temperature-pressure substrate growth maps and resolve how vacancy motifs, phase intermixing and seed structure govern polymorph selection. The simulations reproduce key experimental trends, including the prevalence of $\beta_{12}$/$\chi_3$ phases and their temperature-dependent competition, while revealing kinetic pathways that connect metastable nuclei to long-range order. We identify conditions that suppress competing motifs and promote targeted phases, providing actionable synthesis windows. These results establish a predictive framework for directing borophene growth and, more broadly, for controlling polymorphism in low-dimensional materials by coupling atomistic simulation with machine-learning-enabled phase recognition.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a computational framework that couples a reactive machine-learned interatomic potential with grand-canonical Monte Carlo (GCMC) simulations and data-driven structural classification to model borophene nucleation and growth on Ag(111) and Ag(100). It generates temperature-pressure growth maps, examines the influence of vacancy motifs, phase intermixing, and seed structures on polymorph selection, and claims to reproduce key experimental observations such as the dominance of β12 and χ3 phases and their temperature-dependent competition while identifying kinetic pathways and synthesis conditions that favor targeted phases.
Significance. If the underlying ML potential is shown to be accurate, the work would provide a predictive, atomistic route to directing borophene polymorphism that is currently difficult to achieve experimentally. The combination of GCMC with automated phase recognition enables access to length and time scales relevant to extended-layer formation and could serve as a template for other 2D materials where multiple polymorphs compete.
major comments (2)
- [Methods (potential training/validation) and Results (growth maps)] The central claim that the simulations reproduce experimental β12/χ3 prevalence and temperature-dependent competition rests on the unverified accuracy of the reactive ML interatomic potential for B/Ag energetics, diffusion barriers, and substrate interactions. No quantitative validation metrics (energy/force errors vs. DFT, comparison to measured nucleation barriers, or cross-validation on Ag(111) vs. Ag(100)) are supplied in the Methods or Results sections; any systematic bias in relative phase stabilities would invalidate the derived growth maps and synthesis windows.
- [Results (temperature-pressure maps and synthesis windows)] The temperature-pressure maps and identified synthesis windows lack error bars or uncertainty estimates arising from finite sampling in GCMC or from potential inaccuracies. This weakens the assertion that specific conditions suppress competing motifs, as the robustness of the reported phase boundaries cannot be assessed.
minor comments (2)
- [Abstract and Introduction] The abstract and introduction refer to 'data-driven structural classification' without naming the algorithm (e.g., unsupervised clustering, supervised ML model) or the feature set used; this should be stated explicitly for reproducibility.
- [Figure captions] Figure captions for the growth maps should include the precise criteria used to assign β12 vs. χ3 labels and the simulation cell sizes employed.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback highlighting the need for explicit validation and uncertainty quantification. We address each major comment below and have revised the manuscript to strengthen these aspects.
read point-by-point responses
-
Referee: [Methods (potential training/validation) and Results (growth maps)] The central claim that the simulations reproduce experimental β12/χ3 prevalence and temperature-dependent competition rests on the unverified accuracy of the reactive ML interatomic potential for B/Ag energetics, diffusion barriers, and substrate interactions. No quantitative validation metrics (energy/force errors vs. DFT, comparison to measured nucleation barriers, or cross-validation on Ag(111) vs. Ag(100)) are supplied in the Methods or Results sections; any systematic bias in relative phase stabilities would invalidate the derived growth maps and synthesis windows.
Authors: We agree that quantitative validation metrics for the ML potential are necessary to substantiate the central claims. While the potential was trained on extensive DFT configurations and the reproduction of experimental phase trends offers indirect support, the original manuscript did not report explicit error metrics or cross-validation. In the revised version we will add a Methods subsection with energy/force RMSE values versus DFT, training/validation splits, and substrate-specific cross-validation results. Direct comparison to experimental nucleation barriers is not feasible as such data are not available in the literature, but we will clarify this limitation. revision: yes
-
Referee: [Results (temperature-pressure maps and synthesis windows)] The temperature-pressure maps and identified synthesis windows lack error bars or uncertainty estimates arising from finite sampling in GCMC or from potential inaccuracies. This weakens the assertion that specific conditions suppress competing motifs, as the robustness of the reported phase boundaries cannot be assessed.
Authors: We concur that the absence of uncertainty estimates limits assessment of phase-boundary robustness. The original submission did not include statistical errors from GCMC sampling. In the revision we will conduct additional independent GCMC trajectories, report standard errors on the phase fractions, and add error bars (or shaded uncertainty regions) to the temperature-pressure maps, together with a brief discussion of sensitivity to potential inaccuracies. revision: yes
Circularity Check
No circularity: simulations generate independent predictions compared to external experiments
full rationale
The paper's central results are produced by running GCMC simulations with a reactive ML interatomic potential on Ag substrates, followed by data-driven classification of nuclei and phases. These outputs are then compared against independent experimental trends (β12/χ3 prevalence and temperature dependence) rather than being fitted or defined in terms of those trends. No load-bearing step reduces by construction to a self-citation, a fitted parameter renamed as prediction, or an ansatz smuggled via prior work by the same authors. The ML potential's accuracy is an external assumption, not a circularity issue within the derivation chain.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The reactive machine-learned interatomic potential accurately models boron atom interactions with Ag(111) and Ag(100) surfaces during nucleation and growth.
Reference graph
Works this paper leans on
-
[1]
Mannix, X.-F
A. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Al- ducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santi- ago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam, and N. P. Guisinger, Synthe- sis of borophenes: Anisotropic, two-dimensional boron polymorphs, Science350, 1513 (2015)
2015
-
[2]
B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen, and K. Wu, Experimental realization of two-dimensional boron sheets, Nat. Chem 8, 563 (2016)
2016
-
[3]
Tang and S
H. Tang and S. Ismail-Beigi, Novel Precursors for Boron Nanotubes: The Competition of Two-Center and Three- Center Bonding in Boron Sheets, Phys. Rev. Lett.99, 115501 (2007)
2007
-
[4]
X.Wu, J.Dai, Y.Zhao, Z.Zhuo, J.Yang,andX.C.Zeng, Two-dimensional boron monolayer sheets, ACS Nano6, 7443 (2012)
2012
-
[5]
E. S. Penev, S. Bhowmick, A. Sadrzadeh, and B. I. Yakobson, Polymorphism of two-dimensional boron, Nano Lett.12, 2441 (2012)
2012
-
[6]
Mortazavi, M.-Q
B. Mortazavi, M.-Q. Le, T. Rabczuk, and L. F. C. Pereira, Anomalous strain effect on the thermal con- ductivity of borophene: A reactive molecular dynamics study., Phys. E Low-Dimens. Syst. Nanostructures93, 202 (2017)
2017
-
[7]
Z.Pang, X.Qian, Y.Wei,andR.Yang,Super-stretchable borophene, Eur. Phys. Lett.116, 36001 (2016)
2016
-
[8]
Zhang, Y
Z. Zhang, Y. Yang, E. S. Penev, and B. I. Yakobson, Elasticity, Flexibility, and Ideal Strength of Borophenes, Adv. Funct. Mater.27, 1605059 (2017)
2017
-
[9]
Lherbier, A
A. Lherbier, A. R. Botello-Méndez, and J.-C. Charlier, Electronic and optical properties of pristine and oxidized borophene, 2D Mater.3, 045006 (2016)
2016
-
[10]
Huang, S
Y. Huang, S. N. Shirodkar, and B. I. Yakobson, Two- Dimensional Boron Polymorphs for Visible Range Plas- monics: A First-Principles Exploration, J. Am. Chem. Soc.139, 17181 (2017)
2017
-
[11]
Lian, S.-Q
C. Lian, S.-Q. Hu, J. Zhang, C. Cheng, Z. Yuan, S. Gao, and S. Meng, Integrated Plasmonics: Broadband Dirac Plasmons in Borophene, Phys. Rev. Lett.125, 116802 (2020)
2020
-
[12]
L. Z. Liu, S. J. Xiong, and X. L. Wu, Monolayer borophene electrode for effective elimination of both the Schottky barrier and strong electric field effect, Appl. Phys. Lett.109, 061601 (2016)
2016
-
[13]
Jiang, Z
HR. Jiang, Z. Lu, MC. Wu, F. Ciucci, and TS. Zhao, Borophene : A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries, Nano Energy23, 97 (2016)
2016
-
[14]
L. Li, H. Zhang, and X. Cheng, The high hydrogen stor- age capacities of Li-decorated borophene, Comp. Mat. Sci.137, 119 (2017)
2017
-
[15]
Liang, Y
P. Liang, Y. Cao, B. Tai, L. Zhang, H. Shu, F. Li, D. Chao, and X. Du, Is borophene a suitable anode ma- terial for sodium ion battery?, J. Alloys Comp.704, 152 (2017)
2017
-
[16]
D. Rao, L. Zhang, Z. Meng, X. Zhang, Y. Wang, G. Qiao, X. Shen, H. Xia, J. Liu, and R. Lu, Ultrahigh energy storage and ultrafast ion diffusion in borophene-based anodes for rechargeable metal ion batteries, J. Mater. Chem. A5, 2328 (2017)
2017
-
[17]
Zhang, P
L. Zhang, P. Liang, H.-b. Shu, X.-l. Man, F. Li, J. Huang, Q.-m. Dong, and D.-l. Chao, Borophene as Efficient Sulfur Hosts for Lithium–Sulfur Batteries: Suppress- ing Shuttle Effect and Improving Conductivity, J. Phys. Chem. C121, 15549 (2017)
2017
-
[18]
Mannix, Z Zhang, N.P
A. Mannix, Z Zhang, N.P. Guisinger, B.I. Yakobson, and M.C. Hersam, Borophene as a prototype for synthetic 2D materials development., Nat. Nanotech13, 444 (2018)
2018
-
[19]
C. Hou, G. Tai, Z. Wu, and J. Hao, Borophene: Current Status, Challenges and Opportunities, ChemPlusChem 85, 2186 (2020). 10
2020
-
[20]
Y. V. Kaneti, D. P. Benu, X. Xu, B. Yuliarto, Y. Ya- mauchi, and D. Golberg, Borophene: Two-dimensional Boron Monolayer: Synthesis, Properties, and Potential Applications, Chem. Rev.122, 1000 (2021)
2021
-
[21]
M. Ou, X. Wang, L. Yu, C. Liu, W. Tao, X. Ji, and L. Mei, The Emergence and Evolution of Borophene, Adv. Sci. , 2001801 (2021)
2021
-
[22]
Kumar, A
A. Kumar, A. S. K. Kumar, G. A. Sundaram, F. M. de Souza, R. K. Gupta, and P. V. Pham, Unlocking the potential of borophene: Recent progress in synthe- sis, properties, andapplications,CoordinationChemistry Reviews523, 216246 (2025)
2025
-
[23]
K. Wang, S. Choyal, J. F. Schultz, J. McKenzie, L. Li, X. Liu, and N. Jiang, Borophene: Synthesis, Chem- istry, and Electronic Properties, ChemPlusChem89, e202400333 (2024)
2024
-
[24]
Wang, T.-Y
Z.-Q. Wang, T.-Y. Lü, H.-Q. Wang, Y. P. Feng, and J.- C. Zheng, Review of borophene and its potential appli- cations, Front. Phys.14, 33403 (2019)
2019
-
[25]
G.J.Adekoya, O.C.Adekoya, M.Muloiwa, E.R.Sadiku, W.K.Kupolati,andY.Hamam,AdvancesInBorophene: Synthesis, Tunable Properties, and Energy Storage Ap- plications, Small20, 2403656 (2024)
2024
-
[26]
G. H. Gupta, S. Kadakia, D. Agiwal, T. Keshari, and S. Kumar, Borophene nanomaterials: Synthesis and ap- plications in biosensors, Mater. Adv.5, 1803 (2024)
2024
-
[27]
N. R. Innis, C. Marichy, C. Journet, and C. Bousige, Borophene bottom-up syntheses: A critical review, 2D Mater.12, 022005 (2025)
2025
-
[28]
W. Li, L. Kong, C. Chen, J. Gou, S. Sheng, W. Zhang, H. Li, L. Chen, P. Cheng, and K. Wu, Experimental realization of honeycomb borophene, Sci. Bull.63, 282 (2018)
2018
-
[29]
N. A. Vinogradov, A. Lyalin, T. Taketsugu, A. S. Vino- gradov, and A. Preobrajenski, Single-Phase Borophene on Ir(111): Formation, Structure, and Decoupling from the Support, ACS Nano13, 14511 (2019)
2019
-
[30]
Zhong, J
Q. Zhong, J. Zhang, P. Cheng, B. Feng, W. Li, S. Sheng, H. Li, S. Meng, L. Chen, and K. Wu, Metastable phases of 2D boron sheets on Ag(111), J Phys Cond Mat29, 095002 (2017)
2017
-
[31]
Zhong, L
Q. Zhong, L. Kong, J. Gou, W. Li, S. Sheng, S. Yang, P. Cheng, H. Li, K. Wu, and L. Chen, Synthesis of borophene nanoribbons on Ag(110) surface, Phys Rev Mater.1, 021001 (2017)
2017
-
[32]
Kiraly, X
B. Kiraly, X. Liu, L. Wang, Z. Zhang, A. J. Mannix, B. L. Fisher, B. I. Yakobson, M. C. Hersam, and N. P. Guisinger, Borophene Synthesis on Au(111), ACS Nano 13, 3816 (2019)
2019
-
[33]
Mazaheri, M
A. Mazaheri, M. Javadi, and Y. Abdi, Chemical Vapor DepositionofTwo-DimensionalBoronSheetsbyThermal Decomposition of Diborane, ACS Appl. Mater. Interfaces 13, 8844 (2021)
2021
-
[34]
R. Wu, I. K. Drozdov, S. Eltinge, P. Zahl, S. Ismail-Beigi, I. Božović, and A. Gozar, Large-area single-crystal sheets of borophene on Cu (111) surfaces, Nat. Nanotech14, 44 (2019)
2019
-
[35]
R. Wu, S. Eltinge, I. K. Drozdov, A. Gozar, P. Zahl, J. T. Sadowski, S. Ismail-Beigi, and I. Božović, Micrometre- scale single-crystalline borophene on a square-lattice Cu(100) surface, Nat. Chem.14, 377 (2022)
2022
-
[36]
G. Tai, T. Hu, Y. Zhou, X. Wang, J. Kong, T. Zeng, Y. You, and Q. Wang, Synthesis of Atomically Thin Boron Films on Copper Foils, Angew. Chem. Int. Ed. 54, 15473 (2015)
2015
-
[37]
Sutter and E
P. Sutter and E. Sutter, Large-Scale Layer-by-Layer Syn- thesis of Borophene on Ru(0001), Chem. Mater.33, 8838 (2021)
2021
-
[38]
Radatović, V.Jadriško, S.Kamal, M.Kralj, D.Novko, N
B. Radatović, V.Jadriško, S.Kamal, M.Kralj, D.Novko, N. Vujičić, and M. Petrović, Macroscopic Single-Phase Monolayer Borophene on Arbitrary Substrates, ACS Appl. Mater. Interfaces14, 21727 (2022)
2022
-
[39]
K. M. Omambac, M. Petrović, P. Bampoulis, C. Brand, M. A. Kriegel, P. Dreher, D. Janoschka, U. Hagemann, N. Hartmann, P. Valerius, T. Michely, F. J. Meyer zu Heringdorf, and M. Horn-von Hoegen, Segregation- Enhanced Epitaxy of Borophene on Ir(111) by Thermal Decomposition of Borazine, ACS Nano15, 7421 (2021)
2021
-
[40]
M. G. Cuxart, K. Seufert, V. Chesnyak, W. A. Waqas, A. Robert, M.-L. Bocquet, G. S. Duesberg, H. Sachdev, and W. Auwärter, Borophenes made easy, Sci. Adv.7, eabk1490 (2021)
2021
-
[41]
Sheng, J.-B
S. Sheng, J.-B. Wu, X. Cong, Q. Zhong, W. Li, W. Hu, J. Gou, P. Cheng, P.-H. Tan, L. Chen, and et al., Raman Spectroscopy of Two-Dimensional Borophene Sheets, ACS Nano13, 4133 (2019)
2019
-
[42]
Z. Wu, X. Liang, Y. Liu, M. Xu, R. Zhu, and G. Tai, Synthesis and Anisotropic Memristive Behavior of Borophene Nanosheets, Angew. Chem. Int. Ed.64, e202416041 (2025)
2025
-
[43]
X. Liu, Z. Zhang, L. Wang, B. I. Yakobson, and M. C. Hersam, Intermixing and periodic self-assembly of borophene line defects., Nat. Mater17, 783 (2018)
2018
-
[44]
W. Li, K. Wu, and L. Chen, Epitaxial growth of borophene on substrates, Progress in Surface Science98, 100704 (2023)
2023
-
[45]
Y. Wang, L. Kong, C. Chen, P. Cheng, B. Feng, K. Wu, and L. Chen, Realization of Regular-Mixed Quasi-1D Borophene Chains with Long-Range Order, Adv. Mater. 32, 2005128 (2020)
2020
-
[46]
A. K. Niessen and F. R. De Boer, The enthalpy of for- mation of solid borides, carbides, nitrides, silicides and phosphides of transition and noble metals, Journal of the Less Common Metals82, 75 (1981)
1981
-
[47]
A. P. Sergeeva, I. A. Popov, Z. A. Piazza, W.-L. Li, C. Romanescu, L.-S. Wang, and A. I. Boldyrev, Under- standing Boron through Size-Selected Clusters: Struc- ture, Chemical Bonding, and Fluxionality, Acc. Chem. Res.47, 1349 (2014)
2014
-
[48]
Kiran, S
B. Kiran, S. Bulusu, H.-J. Zhai, S. Yoo, X. C. Zeng, and L.-S. Wang, Planar-to-tubular structural transition in boron clusters: B 20 as the embryo of single-walled boron nanotubes, Proc. Natl. Acad. Sci. U.S.A.102, 961 (2005)
2005
-
[49]
Li, Y.-F
W.-L. Li, Y.-F. Zhao, H.-S. Hu, J. Li, and L.-S. Wang, B− 30 : A Quasiplanar Chiral Boron Cluster, Angew. Chem. Int. Ed.53, 5540 (2014)
2014
-
[50]
Chen, W.-J
Q. Chen, W.-J. Tian, L.-Y. Feng, H.-G. Lu, Y.-W. Mu, H.-J. Zhai, S.-D. Li, and L.-S. Wang, Planar B38 - and B37 - clusters with a double-hexagonal vacancy: Molecu- lar motifs for borophenes, Nanoscale9, 4550 (2017)
2017
-
[51]
Z. A. Piazza, H.-S. Hu, W.-L. Li, Y.-F. Zhao, J. Li, and L.-S. Wang, Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets, Nat. Comm5, 3113 (2014)
2014
-
[52]
H. Liu, J. Gao, and J. Zhao, From Boron Cluster to Two- Dimensional Boron Sheet on Cu(111) Surface: Growth Mechanism and Hole Formation, Sci Rep3, 3238 (2013). 11
2013
-
[53]
S. Xu, Y. Zhao, J. Liao, X. Yang, and H. Xu, The nucle- ation and growth of borophene on the Ag (111) surface, Nano Res.9, 2616 (2016)
2016
-
[54]
H. Shu, F. Li, P. Liang, and X. Chen, Unveiling the atomic structure and electronic properties of atomically thin boron sheets on an Ag(111) surface, Nanoscale8, 16284 (2016)
2016
-
[55]
M. Yu, Y. Gui, Z. Zhao, J. Li, X. Guo, and Z. Zhang, Mechanism for Borophene Phase Transition on Sub- strate, ACS Nano 10.1021/acsnano.5c17811 (2026)
-
[56]
Mignon, A.-R
P. Mignon, A.-R. Allouche, N. R. Innis, and C. Bousige, Neural Network Approach for a Rapid Prediction of Metal-Supported Borophene Properties, J. Am. Chem. Soc.145, 27857 (2023)
2023
-
[57]
Bousige, A.-A
C. Bousige, A.-A. Delenda, A.-R. Allouche, and P. Mignon, A Portable Data Set for Borophene Growth Modeling with Reactive Neural Network Potentials, J. Phys. Chem. C129, 18760 (2025)
2025
-
[58]
G. P. Campbell, A. J. Mannix, J. D. Emery, T.-L. Lee, N. P. Guisinger, M. C. Hersam, and M. J. Bedzyk, Re- solving the Chemically Discrete Structure of Synthetic Borophene Polymorphs, Nano Lett.18, 2816 (2018)
2018
-
[59]
Q. Gao, W. Li, C. Chen, K. Wu, Z. Hu, and L. Chen, Substrate-Induced One-Dimensional Borophene-Silver Hybridization, J. Am. Chem. Soc.148, 23636 (2026)
2026
-
[60]
Bousige, BoroML (2026)
C. Bousige, BoroML (2026)
2026
-
[61]
Behler and M
J. Behler and M. Parrinello, Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces, Phys. Rev. Lett.98, 146401 (2007)
2007
-
[62]
Singraber, T
A. Singraber, T. Morawietz, J. Behler, and C. Dellago, Parallel Multistream Training of High-Dimensional Neu- ral Network Potentials, J. Chem. Theory Comput.15, 3075 (2019)
2019
-
[63]
Singraber, J
A. Singraber, J. Behler, and C. Dellago, Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials, J. Chem. Theory Comput.15, 1827 (2019)
2019
-
[64]
A. Singraber, M. Bircher, S. Reeve, D. W. H. Swenson, J. Lauret, and P. David, CompPhysVienna/n2p2: Ver- sion 2.1.4, Zenodo 10.5281/zenodo.4750573 (2021)
-
[65]
Behler, Atom-centered symmetry functions for con- structing high-dimensional neural network potentials, J
J. Behler, Atom-centered symmetry functions for con- structing high-dimensional neural network potentials, J. Chem. Phys.134, 074106 (2011)
2011
-
[66]
Plimpton, Fast parallel algorithms for short-range molecular dynamics, J
S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comp. Phys.117, 1 (1995)
1995
-
[67]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott, and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Comm...
2022
-
[68]
Frenkel and B
D. Frenkel and B. Smit,Understanding Molecular Simu- lation(2002)
2002
-
[69]
Bradski, Opencv/opencv-python, OpenCV (2026)
G. Bradski, Opencv/opencv-python, OpenCV (2026)
2026
-
[70]
Becker, E
S. Becker, E. Devijver, R. Molinier, and N. Jakse, Unsu- pervised topological learning for identification of atomic structures, Phys. Rev. E105, 045304 (2022)
2022
-
[71]
J.Furstoss, C.R.Salazar, P.Carrez, P.Hirel,andJ.Lam, All-around local structure classification with supervised learning: The example of crystal phases and dislocations in complex oxides, Computer Physics Communications 309, 109480 (2025)
2025
-
[72]
A. M. Goryaeva, C. Lapointe, C. Dai, J. Dérès, J.- B. Maillet, and M.-C. Marinica, Reinforcing materials modelling by encoding the structures of defects in crys- talline solids into distortion scores, Nat Commun11, 4691 (2020)
2020
-
[73]
Huo and M
H. Huo and M. Rupp, Unified representation of molecules and crystals for machine learning, Mach. Learn.: Sci. Technol.3, 045017 (2022)
2022
-
[74]
Himanen, M
L. Himanen, M. O. J. Jäger, E. V. Morooka, F. Fed- erici Canova, Y. S. Ranawat, D. Z. Gao, P. Rinke, and A. S. Foster, DScribe: Library of descriptors for machine learning in materials science, Computer Physics Commu- nications247, 106949 (2020)
2020
-
[75]
A. P. Bartók, R. Kondor, and G. Csányi, On representing chemical environments, Phys. Rev. B87, 184115 (2013)
2013
-
[76]
P. J. Steinhardt, D. R. Nelson, and M. Ronchetti, Bond- orientational order in liquids and glasses, Phys. Rev. B 28, 784 (1983)
1983
-
[77]
Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys
R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys. Rev. B99, 014104 (2019)
2019
-
[78]
Lysogorskiy, C
Y. Lysogorskiy, C. van der Oord, A. Bochkarev, S. Menon, M. Rinaldi, T. Hammerschmidt, M. Mrovec, A. Thompson, G. Csányi, C. Ortner, and R. Drautz, Per- formant implementation of the atomic cluster expansion (PACE) and application to copper and silicon, npj Com- put Mater7, 97 (2021)
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.