Quantum computing for accurate large-scale electronic-structure calculations: DFT-embedded, post-processed quantum-selected configuration interaction
Pith reviewed 2026-06-27 23:22 UTC · model grok-4.3
The pith
A multilevel embedding framework uses quantum-selected configuration interaction to reach ~1 kcal/mol accuracy for large-system reactions on partial quantum hardware.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
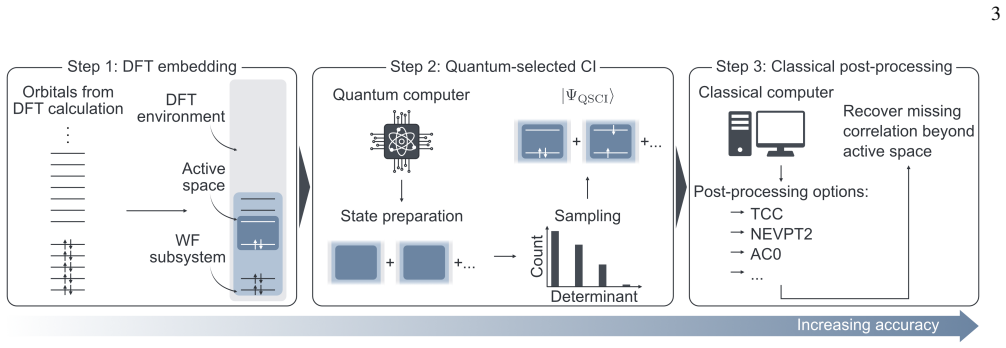
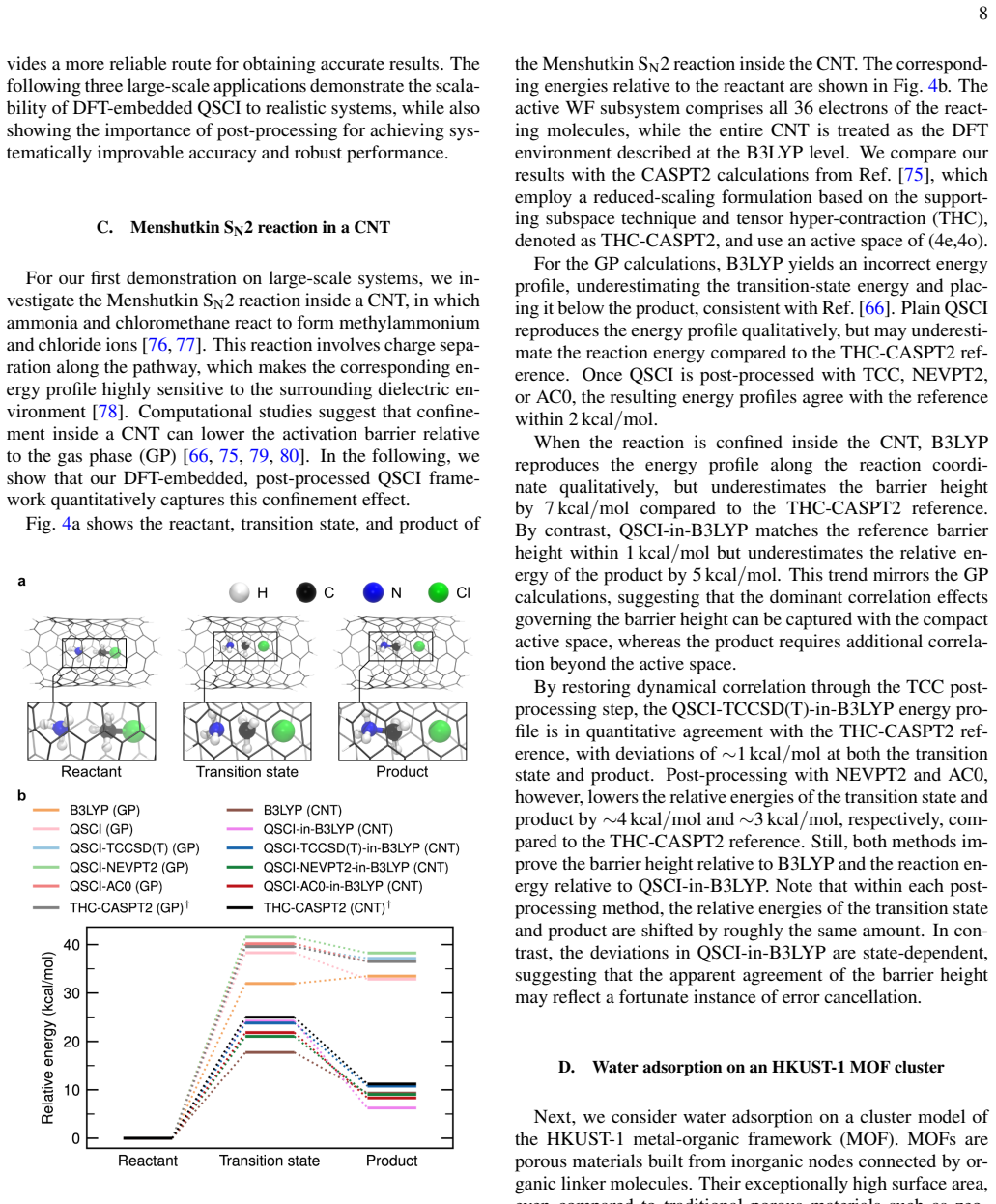
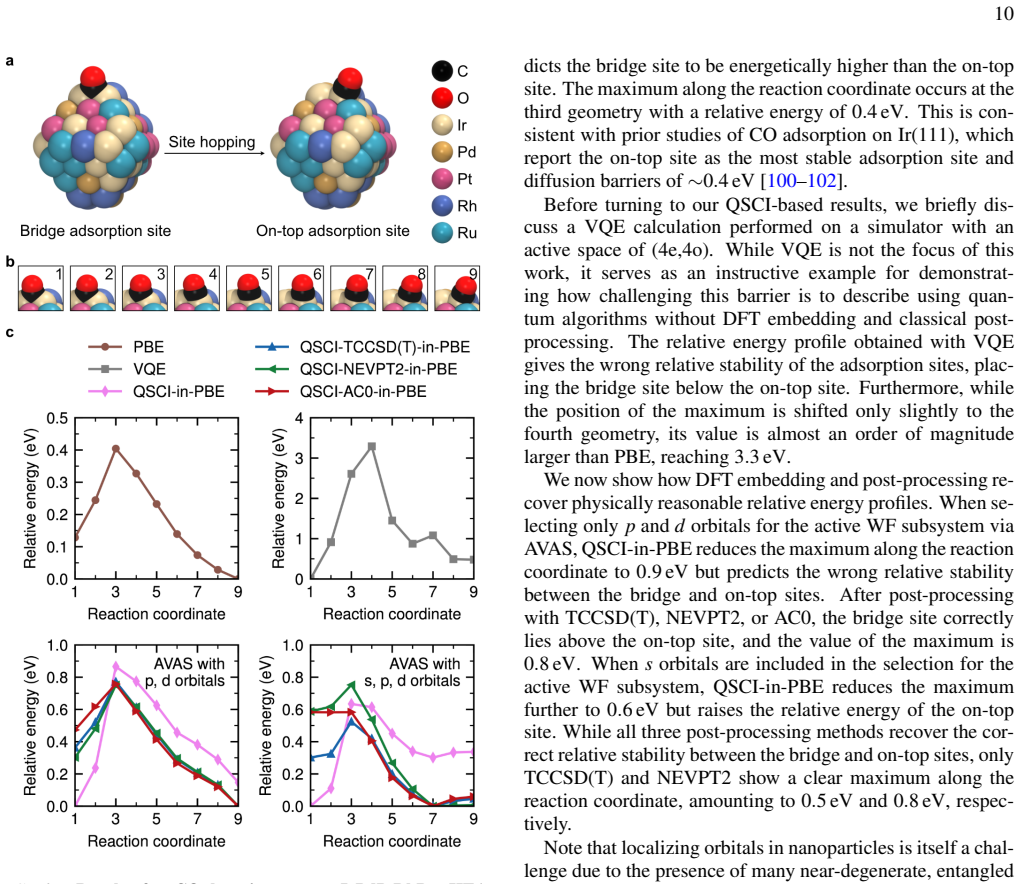
The paper establishes that a DFT-embedded, post-processed quantum-selected configuration interaction framework enables accurate large-scale electronic-structure calculations by letting a quantum computer treat the active space, classical methods recover outer correlation, and density functional theory describe the environment, with the approach demonstrated by ~1 kcal/mol agreement for the Menshutkin SN2 reaction inside a carbon nanotube on a subset of a 144-qubit device.
What carries the argument
The quantum-selected configuration interaction (qSCI) sampling algorithm, which selects configurations from the quantum device to bridge the active-space quantum treatment and classical post-processing inside the DFT embedding.
If this is right
- The framework applies to organic, metal-organic, and metallic systems for computing bond dissociation energies, adsorption energies, and reaction barriers.
- Calculations remain feasible when only a subset of qubits from a 144-qubit superconducting processor is available.
- Chemical accuracy near 1 kcal/mol is reached for a confined SN2 reaction without quantum treatment of every electron.
- Hybrid quantum-classical methods become viable for systems whose size exceeds direct quantum simulation capacity.
Where Pith is reading between the lines
- The same embedding structure could be tested on other confined or heterogeneous reaction environments to check transferability.
- If the qSCI bridge remains reliable, the approach may combine with alternative classical post-processing methods without altering the quantum step.
- Hardware with modestly more qubits would allow larger active spaces while retaining the same DFT outer layer.
Load-bearing premise
The sampling-based quantum-selected configuration interaction algorithm accurately bridges the quantum treatment of the active space and the classical post-processing of the surrounding region within the DFT-embedded framework.
What would settle it
A full classical reference calculation of the Menshutkin SN2 reaction barrier inside the carbon nanotube that differs from the hybrid result by more than 1 kcal/mol.
Figures
read the original abstract
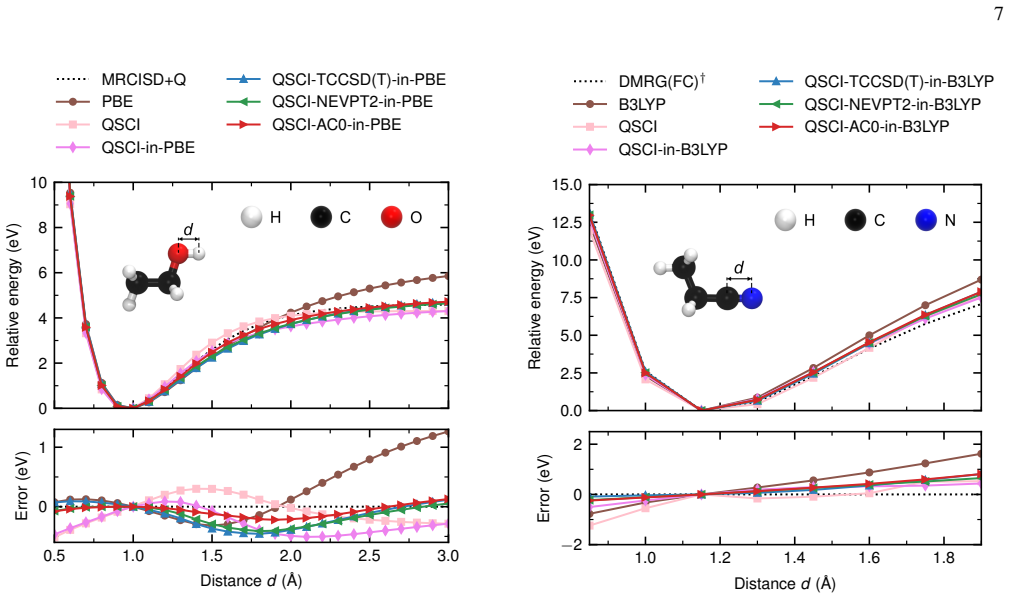
We present a multilevel embedding framework for quantum chemistry calculations on a quantum computer. In our framework, a quantum algorithm treats the strongly correlated active space, while a high-level wave-function method such as coupled cluster theory or multireference perturbation theory recovers the remaining correlation in the surrounding region. A sampling-based quantum algorithm, quantum-selected configuration interaction, bridges the quantum and classical treatments. The entire calculation is embedded in a low-cost density functional theory description of the surrounding environment using Manby's projection technique. We apply the framework to organic, metal-organic, and metallic systems, computing bond dissociation energies, adsorption energies, and reaction barriers using only the subset of qubits of a 144-qubit superconducting quantum computer at the University of Osaka and achieving $\sim$1 kcal/mol agreement with classical references for a Menshutkin $\mathrm{S_N2}$ reaction inside a carbon nanotube. Our results may open the way to quantitatively reliable quantum-classical hybrid calculations for large-scale chemical systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a multilevel embedding framework for quantum chemistry on quantum computers: a quantum algorithm (quantum-selected configuration interaction, QS-CI) treats the strongly correlated active space on a subset of qubits from a 144-qubit superconducting device, classical high-level wavefunction methods (CC or MRPT) recover remaining correlation in the surrounding region, and the whole system is embedded in a DFT description via Manby's projection technique. The framework is applied to bond dissociation, adsorption, and reaction barriers in organic, metal-organic, and metallic systems, with the central numerical result being ~1 kcal/mol agreement with classical references for a Menshutkin SN2 barrier inside a carbon nanotube.
Significance. If the accuracy and bridging claims hold after validation, the work would constitute a meaningful step toward hybrid quantum-classical calculations on chemically relevant scales by partitioning correlation treatment across quantum hardware, classical post-processing, and DFT embedding. The demonstration on real superconducting hardware with only a qubit subset is a concrete strength, as is the parameter-free character of the embedding once the active space is chosen.
major comments (2)
- [Abstract] Abstract and results presentation: the central claim of ∼1 kcal/mol agreement for the Menshutkin SN2 reaction supplies neither error bars on the embedded energies, convergence data with respect to QS-CI sampling shots or active-space size, nor multiple independent classical benchmarks, so the numerical reliability cannot be assessed from the reported information.
- [Methods / Results (bridging algorithm)] QS-CI bridging step: because the method relies on stochastic sampling of configurations from quantum measurements to supply the active-space correlation that is then augmented by classical CC/MRPT inside the DFT embedding, the absence of direct numerical comparisons of QS-CI energies to exact full-CI or DMRG benchmarks on the same active spaces used for the nanotube reaction leaves open the possibility that sampling bias propagates into the final barrier height.
Simulated Author's Rebuttal
We are grateful to the referee for their thorough review and valuable suggestions. We have carefully considered each comment and provide point-by-point responses below. Revisions have been made to improve the clarity and completeness of the numerical results presentation.
read point-by-point responses
-
Referee: [Abstract] Abstract and results presentation: the central claim of ∼1 kcal/mol agreement for the Menshutkin SN2 reaction supplies neither error bars on the embedded energies, convergence data with respect to QS-CI sampling shots or active-space size, nor multiple independent classical benchmarks, so the numerical reliability cannot be assessed from the reported information.
Authors: We agree that additional details on the numerical reliability would strengthen the presentation. In the revised manuscript, we will include error estimates derived from the QS-CI sampling procedure in the abstract and main text. Convergence with respect to the number of shots and active-space size will be documented in the supplementary information. The main text already employs multiple classical methods (including CCSD(T) and MRPT) as benchmarks for the embedded energies; we will make this explicit in a dedicated paragraph. revision: yes
-
Referee: [Methods / Results (bridging algorithm)] QS-CI bridging step: because the method relies on stochastic sampling of configurations from quantum measurements to supply the active-space correlation that is then augmented by classical CC/MRPT inside the DFT embedding, the absence of direct numerical comparisons of QS-CI energies to exact full-CI or DMRG benchmarks on the same active spaces used for the nanotube reaction leaves open the possibility that sampling bias propagates into the final barrier height.
Authors: We acknowledge the referee's concern regarding potential sampling bias. Direct full-CI or DMRG calculations on the active spaces employed for the nanotube system are not feasible with classical methods due to their size. However, the QS-CI algorithm has been extensively benchmarked against exact methods on smaller active spaces in our earlier work. To address this point, we will add a discussion in the revised manuscript explaining the validation strategy and why the hybrid approach mitigates bias through classical post-processing. We believe this provides sufficient assurance for the reported accuracy. revision: partial
Circularity Check
No circularity; external benchmarking against classical references
full rationale
The derivation chain relies on a multilevel embedding framework where QS-CI bridges active-space quantum treatment to classical post-processing within DFT embedding. The central claims (e.g., ~1 kcal/mol agreement for the Menshutkin SN2 barrier) are validated by direct numerical comparison to independent classical references (CC, MRPT, etc.) rather than being defined in terms of fitted outputs or self-citations. No self-definitional equations, fitted-input predictions, or load-bearing self-citation chains appear in the abstract or description; the method is presented as a hybrid whose accuracy is externally falsifiable.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Manby's projection technique provides an accurate low-cost DFT embedding of the active region.
- domain assumption The quantum-selected configuration interaction sampling converges to the correct active-space correlation energy.
Forward citations
Cited by 1 Pith paper
-
QuBE/Qubex: an integrated hardware-software system for superconducting qubit experiments with broadband control
QuBE/Qubex combines broadband microwave hardware with open-source pulse-level software for scalable superconducting qubit experiments, validated on a 64-qubit chip with 98.34% two-qubit gate fidelity.
Reference graph
Works this paper leans on
-
[1]
Projection-based WF-in-DFT embedding In DFT, the energy of a system is expressed as a functional of its total electron densityρ total. When the electron density of the total system is divided into the densityρ A of an active subsystem A and the densityρ B of an environment B, ρtotal =ρ A +ρ B ,(1) the total energyE[ρ total]can be correspondingly partition...
-
[2]
FCI yields the exact ground state within the chosen orbital basis but is intractable for large systems due to its combinatorially increasing computational cost
Quantum-selected configuration interaction (QSCI) In the full configuration interaction (FCI) method, the wave function is written as a linear combination of all Slater deter- minants that can be formed from the chosen orbital space, |ΨFCI⟩ = N ∑ i=1 ci |Φi⟩ ,(9) where |Φi⟩ is theith Slater determinant,c i the corresponding CI coefficient, andNthe total n...
-
[3]
tailored
Post-processing using TCC In the conventional coupled-cluster (CC) method, the wave function is written as |ΨCC⟩=e ˆT |Ψ0⟩,(15) where|Ψ 0⟩is the reference Slater determinant and ˆTthe clus- ter operator. For single-reference systems near equilibrium, CCSD(T) is widely regarded as the gold standard because it provides a highly accurate treatment of dynamic...
-
[4]
Here, it is applied as a post-processing step on top of the QSCI wave function, using the strongly contracted variant
Post-processing using NEVPT2 NEVPT2 is a multireference perturbation theory for recov- ering dynamical correlation from an active-space reference [40–42]. Here, it is applied as a post-processing step on top of the QSCI wave function, using the strongly contracted variant. As a perturbation theory, NEVPT2 is formulated by parti- tioning the Hamiltonian as...
-
[5]
Post-processing using AC0 AC0 is based on an adiabatic connection (AC) that interpo- lates between a chosen reference Hamiltonian and the fully in- teracting electronic Hamiltonian [47]. Compared to represen- tative multireference perturbation methods such as CASPT2 and NEVPT2, AC0 does not require reduced density matri- ces higher than second order, maki...
-
[6]
To account for spin symmetry when constructing the effective Hamiltonian from the sampled determinants (eqn
and QURI Parts OQTOPUS [56, 57] were employed to interface the quantum and classical computations and access the quantum device via cloud-based services. To account for spin symmetry when constructing the effective Hamiltonian from the sampled determinants (eqn. (13)), the union of the α- andβ-spin configuration sets was formed by adding con- figurations ...
-
[7]
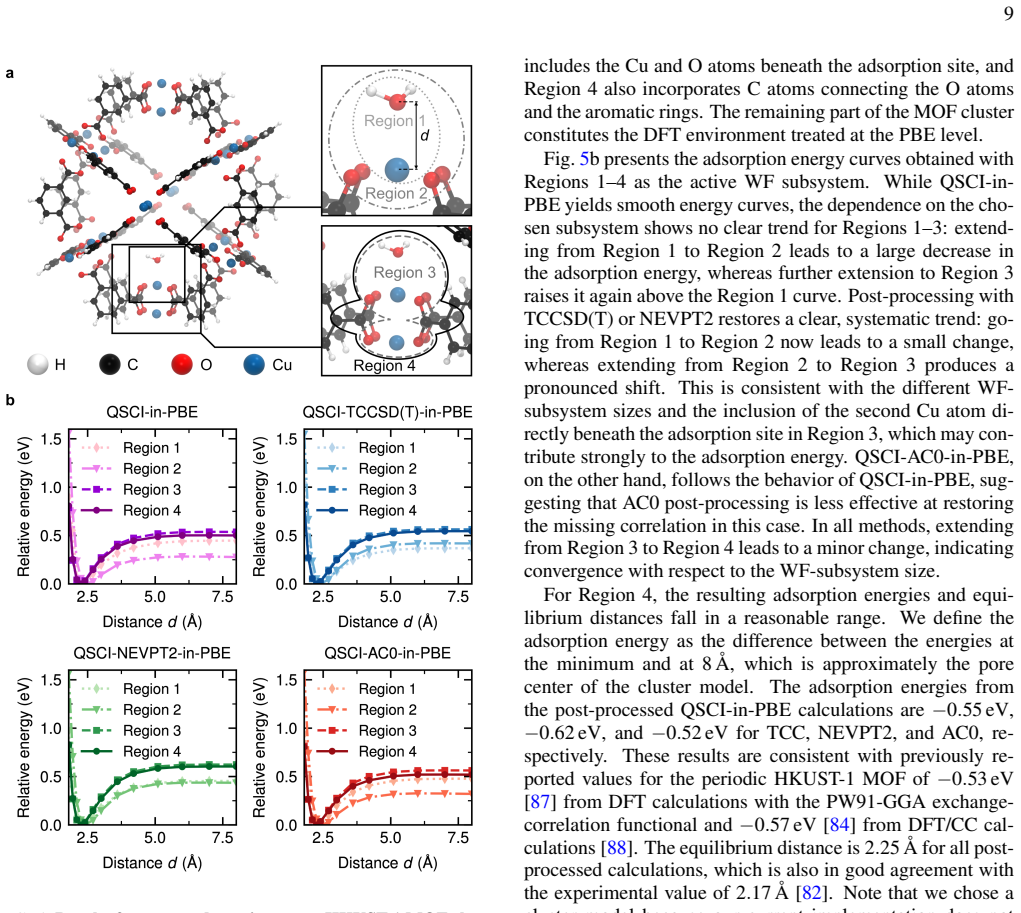
The equilibrium distance is 2.25 Å for all post- processed calculations, which is also in good agreement with the experimental value of 2.17 Å [82]
from DFT calculations with the PW91-GGA exchange- correlation functional and−0.57 eV [84] from DFT/CC cal- culations [88]. The equilibrium distance is 2.25 Å for all post- processed calculations, which is also in good agreement with the experimental value of 2.17 Å [82]. Note that we chose a cluster model because our current implementation does not yet su...
- [8]
-
[9]
AbuGhanem, IBM quantum computers: Evolution, perfor- mance, and future directions, J
M. AbuGhanem, IBM quantum computers: Evolution, perfor- mance, and future directions, J. Supercomput.81, 687 (2025)
2025
-
[10]
A. Ransford, M. S. Allman, J. Arkinstall, J. P. Campora, S. F. Cooper, R. D. Delaney, J. M. Dreiling, B. Estey, C. Fig- gatt, A. Hall, A. A. Husain, A. Isanaka, C. J. Kennedy, N. Kotibhaskar, I. S. Madjarov, K. Mayer, A. R. Milne, A. J. Park, A. P. Reed, R. Ancona, M. P. Andersen, P. Andres- Martinez, W. Angenent, L. Argueta, B. Arkin, L. Ascar- runz, W. ...
Pith/arXiv arXiv 2025
-
[11]
Wintersperger, F
K. Wintersperger, F. Dommert, T. Ehmer, A. Hoursanov, J. Klepsch, W. Mauerer, G. Reuber, T. Strohm, M. Yin, and S. Luber, Neutral atom quantum computing hardware: Perfor- mance and end-user perspective, EPJ Quantum Technol.10, 32 (2023)
2023
-
[12]
H. T. Nguyen, P. Krishnan, D. Krishnaswamy, M. Usman, and R. Buyya, Quantum Cloud Computing: A Review, Open Prob- lems, and Future Directions (2024), arXiv:2404.11420 [cs]
arXiv 2024
-
[13]
V . Khinevich and W. Mizukami, Enhancing quantum com- putations with the synergy of auxiliary field quantum Monte Carlo and computational basis tomography (2025), arXiv:2502.20066 [quant-ph]
arXiv 2025
-
[14]
Erhart, Y
L. Erhart, Y . Yoshida, V . Khinevich, and W. Mizukami, Cou- pled cluster method tailored with quantum computing, Phys. Rev. Res.6, 023230 (2024)
2024
- [15]
-
[16]
Y . Yoshida, L. Erhart, T. Murokoshi, R. Nakagawa, C. Mori, T. Miyanaga, T. Mori, and W. Mizukami, Auxiliary-field 12 quantum Monte Carlo method with quantum selected config- uration interaction (2025), arXiv:2502.21081 [quant-ph]
arXiv 2025
-
[17]
K. Kanno, M. Kohda, R. Imai, S. Koh, K. Mitarai, W. Mizukami, and Y . O. Nakagawa, Quantum-Selected Con- figuration Interaction: Classical diagonalization of Hamilto- nians in subspaces selected by quantum computers (2023), arXiv:2302.11320 [quant-ph]
Pith/arXiv arXiv 2023
-
[18]
Peruzzo, J
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’Brien, A variational eigenvalue solver on a photonic quantum processor, Nat. Com- mun.5, 4213 (2014)
2014
-
[19]
Tilly, H
J. Tilly, H. Chen, S. Cao, D. Picozzi, K. Setia, Y . Li, E. Grant, L. Wossnig, I. Rungger, G. H. Booth, and J. Tennyson, The Variational Quantum Eigensolver: A review of methods and best practices, Phys. Rep.986, 1 (2022)
2022
-
[20]
Robledo-Moreno, M
J. Robledo-Moreno, M. Motta, H. Haas, A. Javadi-Abhari, P. Jurcevic, W. Kirby, S. Martiel, K. Sharma, S. Sharma, T. Shirakawa, I. Sitdikov, R.-Y . Sun, K. J. Sung, M. Takita, M. C. Tran, S. Yunoki, and A. Mezzacapo, Chemistry beyond the scale of exact diagonalization on a quantum-centric super- computer, Sci. Adv.11, eadu9991 (2025)
2025
-
[21]
A. A. Saki, S. Barison, B. Fuller, J. R. Garrison, J. R. Glick, C. Johnson, A. Mezzacapo, J. Robledo-Moreno, M. Rossmannek, P. Schweigert, I. Sitdikov, and K. J. Sung, Qiskit addon: Sample-based quantum diagonalization, https://github.com/Qiskit/qiskit-addon-sqd (2024)
2024
-
[22]
Qiskit contributors and collaborators, Qiskit-addon-sqd-hpc, https://github.com/Qiskit/qiskit-addon-sqd-hpc (2025)
2025
-
[23]
Zhang and H
S. Zhang and H. Krakauer, Quantum Monte Carlo Method using Phase-Free Random Walks with Slater Determinants, Phys. Rev. Lett.90, 136401 (2003)
2003
-
[24]
Kinoshita, O
T. Kinoshita, O. Hino, and R. J. Bartlett, Coupled-cluster method tailored by configuration interaction, J. Chem. Phys. 123, 074106 (2005)
2005
-
[25]
Nakano, R
H. Nakano, R. Uchiyama, and K. Hirao, Quasi-degenerate perturbation theory with general multiconfiguration self- consistent field reference functions, J. Comput. Chem.23, 1166 (2002)
2002
-
[26]
Danilov, J
D. Danilov, J. Robledo-Moreno, K. J. Sung, M. Motta, and J. Shee, Enhancing the Accuracy and Efficiency of Sample- Based Quantum Diagonalization with Phaseless Auxiliary- Field Quantum Monte Carlo, J. Chem. Theory Comput.21, 11585 (2025)
2025
-
[27]
Shirai, S.-Y
S. Shirai, S.-Y . Tseng, H. Iwakiri, T. Horiba, H. Hirai, and S. Koh, Enhancing Accuracy of Quantum-Selected Configu- ration Interaction Calculations Using Multireference Perturba- tion Theory: Application to Aromatic Molecules, ACS Omega 10, 39736 (2025)
2025
-
[28]
Y . Yoshida, T. Murokoshi, R. Nakagawa, C. Mori, Y . Katayama, N. Kuroda, S. Furukawa, H. Tagami, and W. Mizukami, Doubling the size of quantum selected con- figuration interaction based on seniority-zero space and its application to QC-QSCI-AFQMC (2026), arXiv:2602.07912 [quant-ph]
arXiv 2026
-
[29]
Raghavachari, G
K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head- Gordon, A fifth-order perturbation comparison of electron cor- relation theories, Chem. Phys. Lett.157, 479 (1989)
1989
-
[30]
Riplinger and F
C. Riplinger and F. Neese, An efficient and near linear scal- ing pair natural orbital based local coupled cluster method, J. Chem. Phys.138, 034106 (2013)
2013
-
[31]
Riplinger, B
C. Riplinger, B. Sandhoefer, A. Hansen, and F. Neese, Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys.139, 134101 (2013)
2013
-
[32]
Knizia and G
G. Knizia and G. K.-L. Chan, Density Matrix Embedding: A Simple Alternative to Dynamical Mean-Field Theory, Phys. Rev. Lett.109, 186404 (2012)
2012
-
[33]
Knizia and G
G. Knizia and G. K.-L. Chan, Density Matrix Embedding: A Strong-Coupling Quantum Embedding Theory, J. Chem. The- ory Comput.9, 1428 (2013)
2013
-
[34]
C. R. Jacob and J. Neugebauer, Subsystem density-functional theory, WIREs Comput. Mol. Sci.4, 325 (2014)
2014
-
[35]
T. A. Wesolowski, S. Shedge, and X. Zhou, Frozen-Density Embedding Strategy for Multilevel Simulations of Electronic Structure, Chem. Rev.115, 5891 (2015)
2015
-
[36]
F. R. Manby, M. Stella, J. D. Goodpaster, and T. F. I. Miller, A Simple, Exact Density-Functional-Theory Embed- ding Scheme, J. Chem. Theory Comput.8, 2564 (2012)
2012
-
[37]
S. J. R. Lee, M. Welborn, F. R. Manby, and T. F. I. Miller, Projection-Based Wavefunction-in-DFT Embedding, Acc. Chem. Res.52, 1359 (2019)
2019
-
[38]
V orwerk, N
C. V orwerk, N. Sheng, M. Govoni, B. Huang, and G. Galli, Quantum embedding theories to simulate condensed systems on quantum computers, Nat. Comput. Sci.2, 424 (2022)
2022
-
[39]
A. Shajan, D. Kaliakin, A. Mitra, J. R. Moreno, Z. Li, M. Motta, C. Johnson, A. A. Saki, S. Das, I. Sitdikov, A. Mez- zacapo, and K. M. M. Jr, Towards quantum-centric simulations of extended molecules: Sample-based quantum diagonaliza- tion enhanced with density matrix embedding theory (2024), arXiv:2411.09861 [quant-ph]
arXiv 2024
-
[40]
Battaglia, M
S. Battaglia, M. Rossmannek, V . V . Rybkin, I. Tavernelli, and J. Hutter, A general framework for active space embedding methods with applications in quantum computing, npj Com- put. Mater.10, 297 (2024)
2024
-
[41]
T. M. Bickley, A. Mingare, T. Weaving, M. W. de la Bastida, S. Wan, M. Nibbi, P. Seitz, A. Ralli, P. J. Love, M. Chung, M. H. Vera, L. Schulz, and P. V . Coveney, Extending quantum computing through subspace, embedding and classical molec- ular dynamics techniques, Digit. Discov.4, 3427 (2025)
2025
-
[42]
K. Yamamoto, R. Masui, T. Nakajima, M. Tsuji, M. Sato, P. Schow, L. Heidemann, M. Burke, P. Seitz, O. J. Backhouse, J. W. Pedersen, J. Children, C. Holliman, N. Lysne, D. Okuno, S. Sivarajah, D. M. Ramo, A. Chernoguzov, and R. Duncan, Quantum-HPC hybrid computation of biomolecular excited- state energies (2026), arXiv:2601.15677 [quant-ph]
arXiv 2026
-
[43]
J. Merz, A. Shajan, D. Kaliakin, F. Liang, Y . Otsuka, T. Shi- rakawa, L. Broers, H. Xu, M. Tsuji, M. Sato, S. Yunoki, R. Wakizaka, Y . Kawashima, J. Doi, T. Itoko, H. Horii, T. Pellegrini, J. R. Moreno, K. J. Sung, E. Fejer, R. Walkup, S. Seelam, and M. Motta, Crossing the 12,000-atom bar- rier with heterogeneous quantum-classical supercomputing: Quantum...
Pith/arXiv arXiv 2026
-
[44]
Lloyd, Universal Quantum Simulators, Science273, 1073 (1996)
S. Lloyd, Universal Quantum Simulators, Science273, 1073 (1996)
1996
-
[45]
Aspuru-Guzik, A
A. Aspuru-Guzik, A. D. Dutoi, P. J. Love, and M. Head- Gordon, Simulated Quantum Computation of Molecular En- ergies, Science309, 1704 (2005)
2005
-
[46]
V . Khinevich, Y . Lee, N. Yoshioka, and W. Mizukami, Quan- tum Power Iteration Unified Using Generalized Quantum Sig- nal Processing (2025), arXiv:2507.11142 [quant-ph]
arXiv 2025
-
[47]
Angeli, R
C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger, and J.-P. Malrieu, Introduction of n-electron valence states for multiref- erence perturbation theory, J. Chem. Phys.114, 10252 (2001)
2001
-
[48]
Angeli, R
C. Angeli, R. Cimiraglia, and J.-P. Malrieu,N-electron va- lence state perturbation theory: A fast implementation of the strongly contracted variant, Chem. Phys. Lett.350, 297 (2001). 13
2001
-
[49]
Angeli, R
C. Angeli, R. Cimiraglia, and J.-P. Malrieu, N-electron valence state perturbation theory: A spinless formulation and an effi- cient implementation of the strongly contracted and of the par- tially contracted variants, J. Chem. Phys.117, 9138 (2002)
2002
-
[50]
Pastorczak and K
E. Pastorczak and K. Pernal, Correlation Energy from the Adiabatic Connection Formalism for Complete Active Space Wave Functions, J. Chem. Theory Comput.14, 3493 (2018)
2018
-
[51]
Svensson, S
M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber, and K. Morokuma, ONIOM: A Multilayered In- tegrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels-Alder Re- actions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition, J. Phys. Chem.100, 19357 (1996)
1996
-
[52]
D. I. Lyakh, V . F. Lotrich, and R. J. Bartlett, The ‘tailored’ CCSD(T) description of the automerization of cyclobutadiene, Chem. Phys. Lett.501, 166 (2011)
2011
-
[53]
K. G. Dyall, The choice of a zeroth-order Hamiltonian for second-order perturbation theory with a complete active space self-consistent-field reference function, J. Chem. Phys.102, 4909 (1995)
1995
-
[54]
Pernal, Electron Correlation from the Adiabatic Connec- tion for Multireference Wave Functions, Phys
K. Pernal, Electron Correlation from the Adiabatic Connec- tion for Multireference Wave Functions, Phys. Rev. Lett.120, 013001 (2018)
2018
-
[55]
Shiozaki, BAGEL: Brilliantly Advanced General Electronic-structure Library, WIREs Comput
T. Shiozaki, BAGEL: Brilliantly Advanced General Electronic-structure Library, WIREs Comput. Mol. Sci. 8, e1331 (2018)
2018
-
[56]
Pipek and P
J. Pipek and P. G. Mezey, A fast intrinsic localization proce- dure applicable for ab initio and semiempirical linear combi- nation of atomic orbital wave functions, J. Chem. Phys.90, 4916 (1989)
1989
-
[57]
E. R. Sayfutyarova, Q. Sun, G. K.-L. Chan, and G. Knizia, Automated Construction of Molecular Active Spaces from Atomic Valence Orbitals, J. Chem. Theory Comput.13, 4063 (2017)
2017
-
[58]
Chemqulacs, https://wmizukami.github.io/chemqulacs/ (2023)
2023
-
[59]
Kandala, A
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, Hardware-efficient varia- tional quantum eigensolver for small molecules and quantum magnets, Nature549, 242 (2017)
2017
-
[60]
S. Bravyi, J. M. Gambetta, A. Mezzacapo, and K. Temme, Ta- pering off qubits to simulate fermionic Hamiltonians (2017), arXiv:1701.08213 [quant-ph]
Pith/arXiv arXiv 2017
-
[61]
P. A. Spring, L. Milanovic, Y . Sunada, S. Wang, A. F. van Loo, S. Tamate, and Y . Nakamura, Fast Multiplexed Superconducting-Qubit Readout with Intrinsic Purcell Filter- ing Using a Multiconductor Transmission Line, PRX Quan- tum6, 020345 (2025)
2025
-
[62]
QURI Parts, https://github.com/QunaSys/quri-sdk (2024)
2024
-
[63]
OQTOPUS, https://github.com/oqtopus-team (2025)
2025
- [64]
-
[65]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma, S. Wouters, and G. K.-L. Chan, PySCF: The Python-based simulations of chemistry framework, WIREs Comput. Mol. Sci.8, e1340 (2018)
2018
-
[66]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, J. J. Eriksen, Y . Gao, S. Guo, J. Hermann, M. R. Hermes, K. Koh, P. Ko- val, S. Lehtola, Z. Li, J. Liu, N. Mardirossian, J. D. McClain, M. Motta, B. Mussard, H. Q. Pham, A. Pulkin, W. Purwanto, P. J. Robinson, E. Ronca, E. R. Sayfutyarova, M. S...
2020
-
[67]
PySCF-AC0, https://github.com/Quantinuum/pyscf-ac0 (2024)
2024
-
[68]
Neese, The ORCA program system, WIREs Comput
F. Neese, The ORCA program system, WIREs Comput. Mol. Sci.2, 73 (2012)
2012
-
[69]
Neese, F
F. Neese, F. Wennmohs, U. Becker, and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys. 152, 224108 (2020)
2020
-
[70]
Neese, The SHARK integral generation and digestion sys- tem, J
F. Neese, The SHARK integral generation and digestion sys- tem, J. Comput. Chem.44, 381 (2023)
2023
-
[71]
Neese, Software Update: The ORCA Program System— Version 6.0, WIREs Comput
F. Neese, Software Update: The ORCA Program System— Version 6.0, WIREs Comput. Mol. Sci.15, e70019 (2025)
2025
-
[72]
Beran, K
P. Beran, K. Pernal, F. Pavoševi ´c, and L. Veis, Projection- Based Density Matrix Renormalization Group in Density Functional Theory Embedding, J. Phys. Chem. Lett.14, 716 (2023)
2023
-
[73]
Claudino and N
D. Claudino and N. J. Mayhall, Automatic Partition of Orbital Spaces Based on Singular Value Decomposition in the Context of Embedding Theories, J. Chem. Theory Comput.15, 1053 (2019)
2019
-
[74]
W. Xue, Z. Zhang, H. Huang, C. Zhong, and D. Mei, Theoreti- cal Insights into the Initial Hydrolytic Breakdown of HKUST- 1, J. Phys. Chem. C124, 1991 (2020)
1991
- [75]
-
[76]
Jónsson, G
H. Jónsson, G. Mills, and K. W. Jacobsen, Nudged elastic band method for finding minimum energy paths of transitions, in Classical and Quantum Dynamics in Condensed Phase Simu- lations(WORLD SCIENTIFIC, 1998) pp. 385–404
1998
-
[77]
Henkelman and H
G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys.113, 9978 (2000)
2000
-
[78]
T. D. Kühne, M. Iannuzzi, M. Del Ben, V . V . Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V . Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bu...
2020
-
[79]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gra- dient Approximation Made Simple, Phys. Rev. Lett.77, 3865 (1996)
1996
-
[80]
Goedecker, M
S. Goedecker, M. Teter, and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B54, 1703 (1996)
1996
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.