Collaborative estimation and evaluation of SARS-CoV-2 variant nowcasting in the United States
Pith reviewed 2026-06-27 20:29 UTC · model grok-4.3
The pith
A simple national baseline model for SARS-CoV-2 variant frequencies performs as well as or better than most individual models submitted to a new US nowcast hub.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
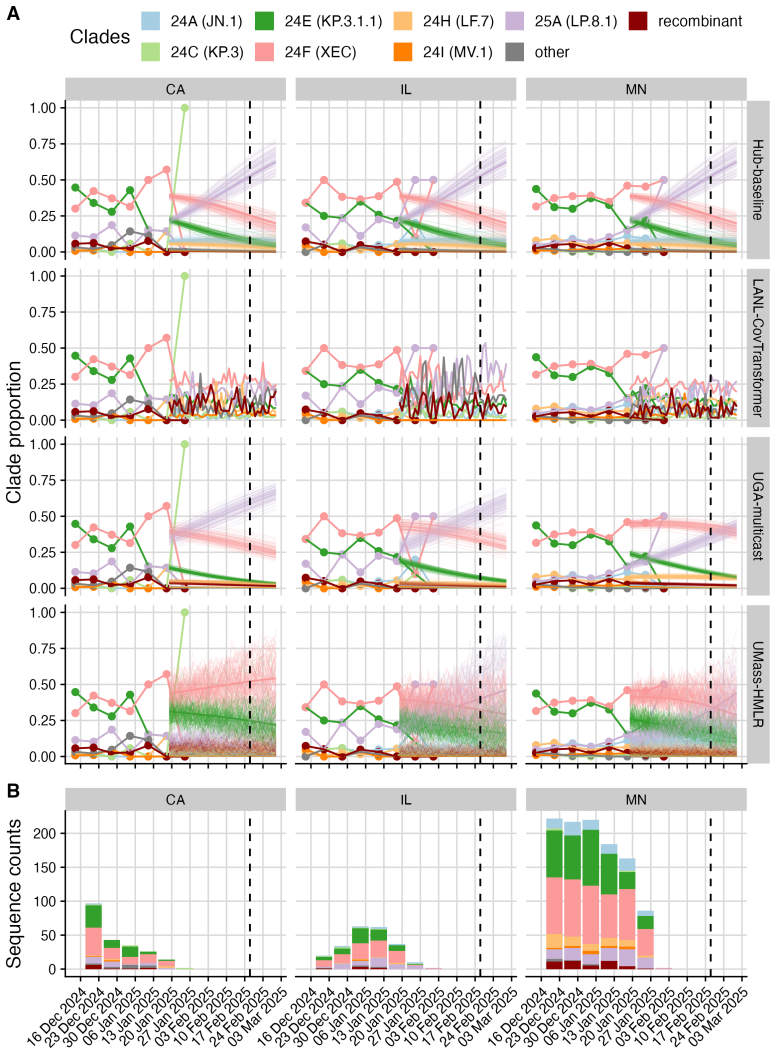
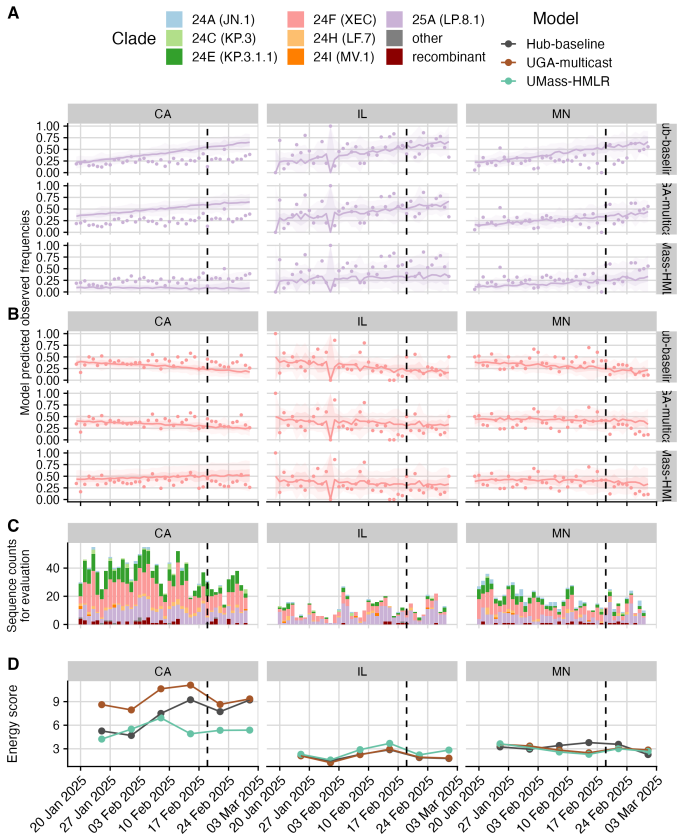
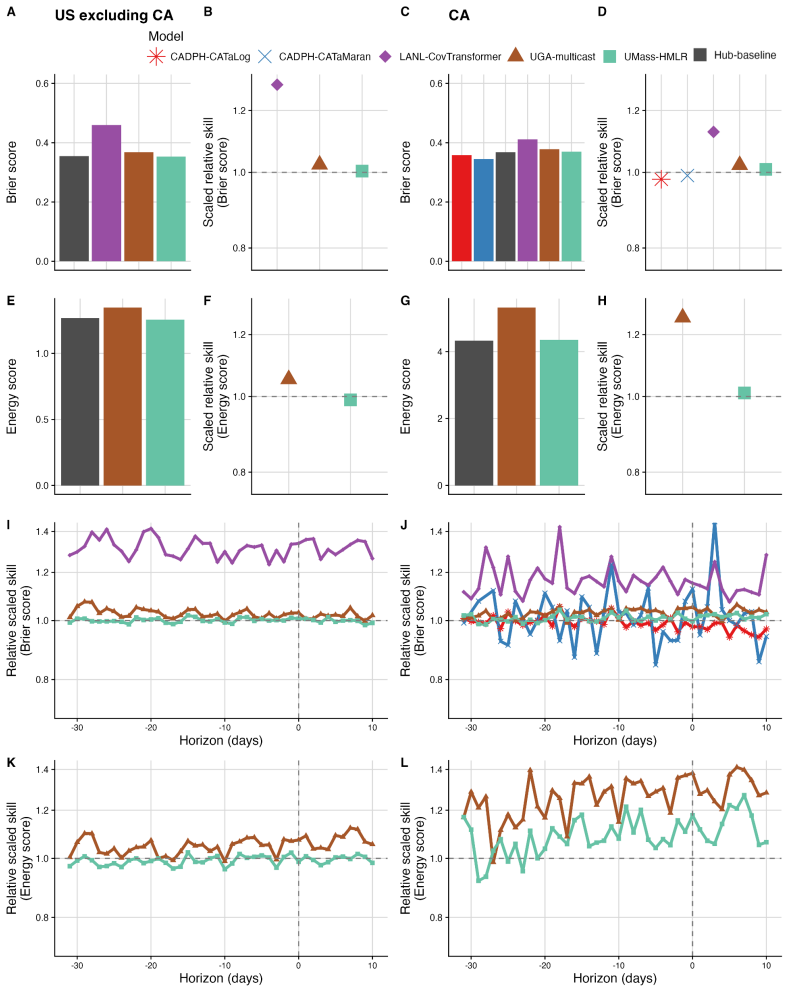
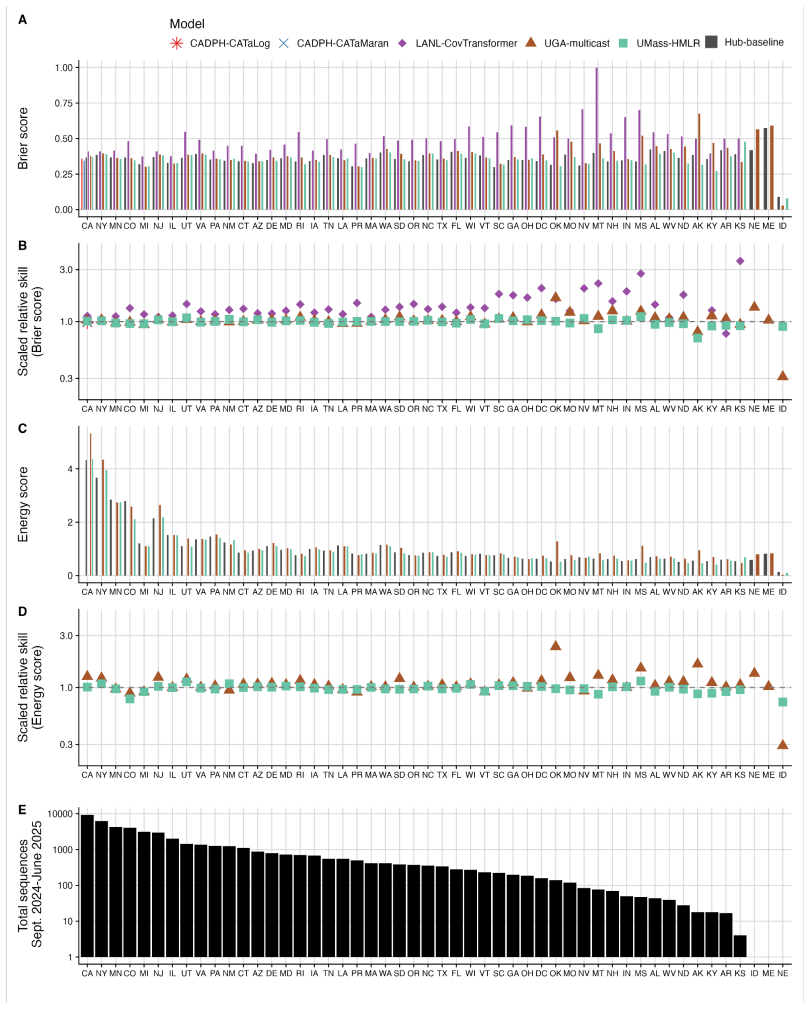
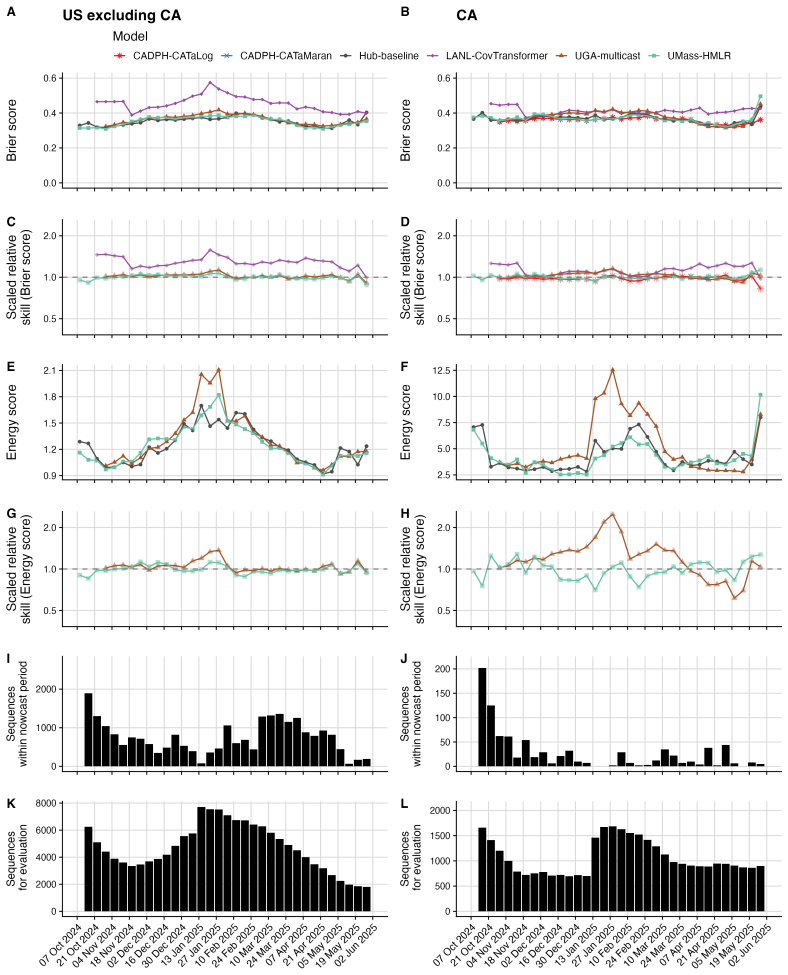
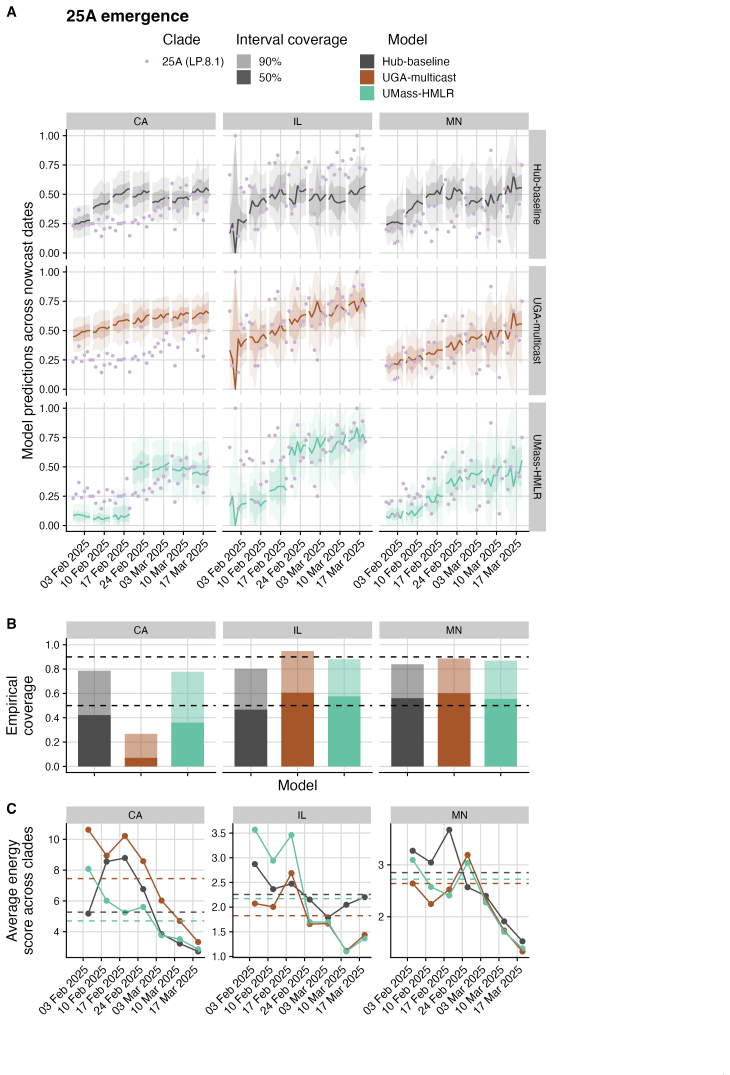
The central claim is that in the Hub's first respiratory virus season, the baseline model pooling sequences across the U.S. performs well overall, with most individual models performing similarly or slightly worse; locations with lower sequencing volumes show greater variability in model performance; and models submitted for a single location outperform those submitted for all locations, potentially due to greater timeliness and magnitude of local data.
What carries the argument
The United States SARS-CoV-2 Variant Nowcast Hub together with its scoring procedures for comparing state-level variant abundance estimates from multiple models against observed sequence data.
If this is right
- The baseline national pooling approach supplies a strong reference that most submitted models do not clearly beat.
- Model performance becomes more variable in states that have lower sequencing volumes.
- Models focused on one location can outperform all-location models because they incorporate more timely or larger local data sets.
- Performance differences may depend on the phase of variant emergence, requiring further targeted checks.
Where Pith is reading between the lines
- For other pathogens with patchy surveillance, national pooling could serve as a default that is hard to beat without extra local effort.
- Low-sequencing states might gain most from added data collection rather than from more complex models.
- The Hub structure could be extended to test whether the same baseline advantage appears for influenza or other respiratory viruses.
Load-bearing premise
The models and evaluation period from the first season provide a representative test of relative performance across different locations and data volumes.
What would settle it
In a later season with new variant dynamics, most individual models consistently and substantially outperform the national baseline across many states with varying sequencing volumes.
Figures
read the original abstract
The ability to estimate and predict pathogen variant dynamics can inform public health responses, including planning for increased transmission or severity, shifts in population immunity, or changes to vaccine or therapeutic effectiveness. The COVID-19 pandemic demonstrated the importance of monitoring SARS-CoV-2 variant evolution through viral genome sequencing, enabling predictive models to estimate variant frequencies in the recent past, present, and short-term future. Collaborative forecasting Hubs provided a valuable way to centralize predictive modeling of epidemiological indicators such as cases, hospitalizations, and deaths during the pandemic; however, none existed for variant dynamics. Here, we discuss the creation of the United States SARS-CoV-2 Variant Nowcast Hub, designed to solicit estimates of the relative abundance of a specified set of SARS-CoV-2 variants at the U.S. state level. We discuss the design decisions and challenges in building the Hub and its scoring procedures. Using submissions from the Hub's first respiratory virus season (nowcast dates October 9th, 2024 to June 4th, 2025), we evaluate five individual models and a baseline model. We found that the baseline model, which pools sequences across the U.S., performs well overall, with most individual models performing similarly or slightly worse. Locations with lower sequencing volumes exhibited greater variability in model performance. Models submitted for a single location outperformed those submitted for all locations, potentially due to greater timeliness and magnitude of local data. Much remains to be investigated regarding relative model performance across different phases of variant emergence, and we conclude by proposing future directions within and beyond this Hub.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript describes the creation of the United States SARS-CoV-2 Variant Nowcast Hub for soliciting state-level estimates of SARS-CoV-2 variant relative abundances. It covers design decisions, challenges, and scoring procedures. Using submissions from the first respiratory virus season (nowcast dates October 9, 2024 to June 4, 2025), the authors evaluate five individual models plus a national-pooling baseline, reporting that the baseline performs well overall while most individual models perform similarly or slightly worse, with greater variability in low-sequencing-volume locations and better performance for location-specific submissions.
Significance. If the evaluation holds, the work establishes a new collaborative infrastructure for variant nowcasting that fills a gap left by prior COVID-19 forecasting hubs. The empirical comparison against external sequence data supplies initial benchmarks and highlights practical effects of sequencing volume and submission scope. Strengths include the open Hub framework and the explicit acknowledgment that further investigation is needed across variant emergence phases; these elements support reproducibility and incremental progress in applied statistical surveillance.
major comments (1)
- [Evaluation / Results] The central performance claims rest on an empirical evaluation whose scoring rules, data exclusion criteria, error handling, and any statistical tests are not described in the abstract and are only alluded to in the provided summary. Without these details (e.g., the precise loss function, weighting by sequencing volume, or handling of zero-count locations), the statement that the baseline “performs well overall” cannot be independently verified and is load-bearing for the paper’s main result.
minor comments (2)
- [Abstract] Abstract: the sentence on scoring procedures could be expanded by one clause naming the primary metric (e.g., weighted absolute error or log-score) to give readers immediate context.
- [Methods] The manuscript would benefit from a concise table listing the five individual models, their submission scopes (single-location vs. all-location), and key methodological differences.
Simulated Author's Rebuttal
We thank the referee for their review. We address the single major comment below and have revised the manuscript accordingly to improve transparency of the evaluation.
read point-by-point responses
-
Referee: [Evaluation / Results] The central performance claims rest on an empirical evaluation whose scoring rules, data exclusion criteria, error handling, and any statistical tests are not described in the abstract and are only alluded to in the provided summary. Without these details (e.g., the precise loss function, weighting by sequencing volume, or handling of zero-count locations), the statement that the baseline “performs well overall” cannot be independently verified and is load-bearing for the paper’s main result.
Authors: We agree that the abstract does not contain these methodological details, which is standard due to length limits, and that the main result would benefit from greater accessibility. The full manuscript describes the scoring rules, data exclusion criteria, error handling, and evaluation approach (including weighting by sequencing volume and treatment of zero-count locations) in the dedicated Scoring and Evaluation sections. No formal statistical tests are used; comparisons are descriptive. To address the concern directly, we have revised the abstract to include a concise statement of the evaluation metric and weighting scheme, and we have added an explicit summary paragraph in the Results section that restates the key aspects of the scoring procedure. These changes allow independent verification of the claim that the baseline performs well overall without altering the underlying analysis. revision: yes
Circularity Check
No significant circularity; empirical evaluation only
full rationale
The paper reports an empirical comparison of nowcasting model submissions against external SARS-CoV-2 sequence data over October 2024–June 2025. No equations, derivations, or first-principles predictions appear; the baseline (national pooling) is a simple, non-fitted reference evaluated directly on held-out observations. No self-citation chains, ansatzes, or fitted-input-as-prediction steps are load-bearing. The central claims rest on observable performance differences by location and model type, making the analysis self-contained.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Genomic sequence data accurately reflect underlying variant frequencies at state level
Reference graph
Works this paper leans on
-
[1]
Fitness models provide accurate short-term forecasts of SARS-CoV-2 variant frequency
Eslam Abousamra, Marlin Figgins, and Trevor Bedford. “Fitness models provide accurate short-term forecasts of SARS-CoV-2 variant frequency”. In:PLOS Computational Biology20.9 (2024), e1012443.doi:10.1371/ journal.pcbi.1012443
2024
-
[2]
Nextclade: clade assignment, mutation calling and quality control for viral genomes
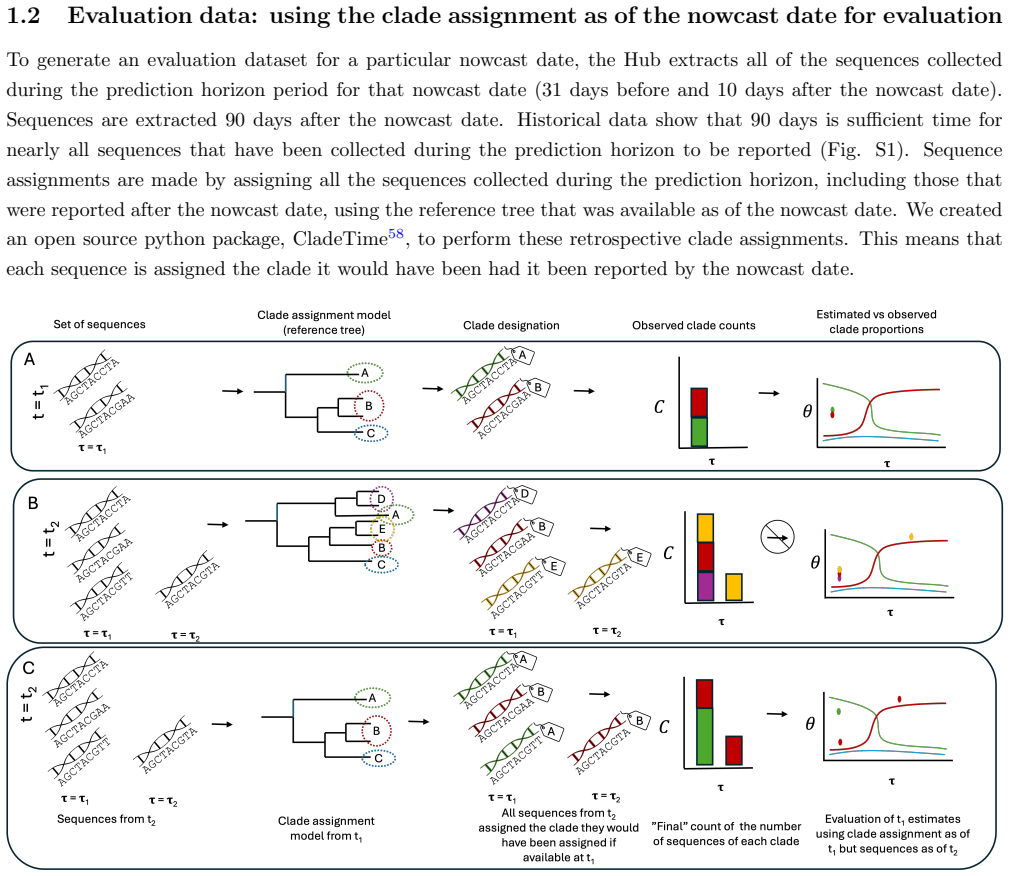
Ivan Aksamentov et al. “Nextclade: clade assignment, mutation calling and quality control for viral genomes”. In:Journal of Open Source Software6.67 (2021), p. 3773.doi:10.21105/joss.03773.url:https://doi. org/10.21105/joss.03773
-
[3]
Nextstrain automates real-time phylodynamic analysis of open data for endemic and emerging pathogens
Kirsty R Andrews et al. “Nextstrain automates real-time phylodynamic analysis of open data for endemic and emerging pathogens”. In:bioRxiv2026.03.23.713807 (2026). Preprint.doi:10.64898/2026.03.23.713807
-
[4]
Molecular Evolution and Epidemiology of Norovirus GII.4 Viruses in the United States
Leslie Barclay et al. “Molecular Evolution and Epidemiology of Norovirus GII.4 Viruses in the United States”. In:The Journal of Infectious Diseases232.4 (Oct. 2025), pp. 933–942.issn: 1537-6613.doi:10.1093/infdis/ jiaf100.url:https://doi.org/10.1093/infdis/jiaf100
-
[5]
“GenBank”
Dennis A Benson et al. “GenBank”. In:Nucleic acids research41.D1 (2012), pp. D36–D42
2012
-
[6]
Binaural room impulse responses recorded with KEMAR in a small meeting room,
Nikos I. Bosse et al.scoringutils: Utilities for Scoring and Assessing Predictions. 2020.doi:10.5281/zenodo. 4618017.url:https://cran.r-project.org/package=scoringutils
-
[7]
Evaluating epidemic forecasts in an interval format
Johannes Bracher et al. “Evaluating epidemic forecasts in an interval format”. In:PLOS Computational Biology17.2 (2021), e1008618.doi:10.1371/journal.pcbi.1008618
-
[8]
Novel (d)PCR assays for influenza A(H5Nx) viruses clade 2.3.4.4b surveillance
Gerhard Buttinger et al. “Novel (d)PCR assays for influenza A(H5Nx) viruses clade 2.3.4.4b surveillance”. In: Eurosurveillance30.33 (2025). Received: 12 Mar 2025; Accepted: 26 Jun 2025, pii=2500183.doi:10.2807/ 1560- 7917.ES.2025.30.33.2500183.url:https://doi.org/10.2807/1560- 7917.ES.2025.30.33. 2500183
-
[9]
Finlay Campbell et al. “Bayesian inference of transmission chains using timing of symptoms, pathogen genomes and contact data”. In:PLOS Computational Biology15.3 (2019), e1006930.doi:10.1371/journal.pcbi. 1006930
-
[10]
GitHub repository
CDC FluSight Team.FluSight Forecast Hub: Repository for Forecasts of Influenza Hospitalizations. GitHub repository. Accessed: 2026-01-12. 2023.url:https://github.com/cdcepi/FluSight-forecast-hub
2026
-
[11]
https://ndc.services.cdc.gov/wp- content/uploads/MMWR Week overview.pdf
Centers for Disease Control and Prevention.MMWR Week Fact Sheet. https://ndc.services.cdc.gov/wp- content/uploads/MMWR Week overview.pdf
-
[12]
cdc.gov/covid/php/variants/variants-and-genomic-surveillance.html
Centers for Disease Control and Prevention.SARS-CoV-2 Variants and Genomic Surveillance.https://www. cdc.gov/covid/php/variants/variants-and-genomic-surveillance.html. Accessed: 2026-01-11. 2024
2026
-
[13]
Accessed: 2026-01-11
Chaoran Chen et al.CoV-Spectrum: Analysis of Globally Shared SARS-CoV-2 Data to Identify and Char- acterize New Variants.https://cov- spectrum.org/explore/United%20Kingdom/AllSamples/Past6M. Accessed: 2026-01-11. 2022. 20
2026
-
[14]
Global landscape of SARS-CoV-2 genomic surveillance and data sharing
Zugen Chen et al. “Global landscape of SARS-CoV-2 genomic surveillance and data sharing”. In:Nature Genetics54 (2022). Published online 28 March 2022, pp. 499–507.doi:10.1038/s41588-022-01033-y
-
[15]
Coordinating collaborative infectious disease modeling projects with the hubverse
Consortium of Infectious Disease Modeling Hubs et al. “Coordinating collaborative infectious disease modeling projects with the hubverse”. In:medRxiv(2025). Preprint.doi:10 . 1101 / 2025 . 10 . 03 . 25337284.url: https://www.medrxiv.org/content/10.1101/2025.10.03.25337284v1
-
[16]
COVID-19 Genomics UK (COG-UK) Consortium.COG-UK Consortium.https : / / www . sanger . ac . uk / collaboration/covid- 19- genomics- uk- cog- uk- consortium/. Accessed: 2026-01-11. Wellcome Sanger Institute, 2020
2026
-
[17]
Estee Y Cramer et al. “Evaluation of individual and ensemble probabilistic forecasts of COVID-19 mortality in the United States”. In:Proceedings of the National Academy of Sciences119.15 (2022), e2113561119.doi: 10.1073/pnas.2113561119
-
[18]
Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic
Alexander Crits-Christoph et al. “Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic”. In:Cell187.19 (2024), 5468–5482.e11.doi:10.1016/j.cell.2024.08.010
-
[19]
Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England
Nicholas G. Davies et al. “Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England”. In:Science372.6538 (2021), eabg3055.doi:10.1126/science.abg3055. eprint:https://www.science. org/doi/pdf/10.1126/science.abg3055.url:https://www.science.org/doi/abs/10.1126/science. abg3055
-
[20]
CovTransformer: A transformer model for SARS-CoV-2 lineage frequency forecasting
Yinan Feng et al. “CovTransformer: A transformer model for SARS-CoV-2 lineage frequency forecasting”. In:Virus Evolution10.1 (2024), veae086.doi:10.1093/ve/veae086.url:https://doi.org/10.1093/ve/ veae086
work page doi:10.1093/ve/veae086.url:https://doi.org/10.1093/ve/ 2024
-
[21]
Marlin D. Figgins and Trevor Bedford.Frequency dynamics predict viral fitness, antigenic relationships and epidemic growth. medRxiv preprint. 2024.doi:10.1101/2024.12.02.24318334.url:https://doi.org/ 10.1101/2024.12.02.24318334
work page doi:10.1101/2024.12.02.24318334.url:https://doi.org/ 2024
-
[23]
Fabr` ıcia Giardina et al. “Methods combining genomic and epidemiological data in the reconstruction of trans- mission trees: a systematic review”. In:Pathogens11.2 (2022), p. 252.doi:10.3390/pathogens11020252
-
[24]
Tilmann Gneiting, Fadoua Balabdaoui, and Adrian E
Tilmann Gneiting and Adrian E. Raftery. “Strictly Proper Scoring Rules, Prediction, and Estimation”. In: Journal of the American Statistical Association102.477 (2007), pp. 359–378.doi:10.1198/016214506000001437
-
[25]
Assessing probabilistic forecasts of multivariate quantities, with an application to ensemble predictions of surface winds
Tilmann Gneiting et al. “Assessing probabilistic forecasts of multivariate quantities, with an application to ensemble predictions of surface winds”. In:Test17 (2008), pp. 211–235
2008
-
[26]
Nextstrain: real-time tracking of pathogen evolution
James Hadfield et al. “Nextstrain: real-time tracking of pathogen evolution”. In:Bioinformatics34.23 (2018), pp. 4121–4123.doi:10.1093/bioinformatics/bty407.url:https://academic.oup.com/bioinformatics/ article/34/23/4121/5001388
work page doi:10.1093/bioinformatics/bty407.url:https://academic.oup.com/bioinformatics/ 2018
-
[27]
org / sars - cov - 2 / forecasts
James Hadfield et al.Nextstrain: SARS-CoV-2 Forecasts.https : / / nextstrain . org / sars - cov - 2 / forecasts. Accessed: 2026-01-16. 2024
2026
-
[28]
James A Hay et al.Evaluation of the epidemiological outlook of the influenza A/H3N2 clade K in England during the 2025-26 season. Preprint.https : / / doi . org / 10 . 5281 / zenodo . 17704679. Nov. 2025.doi: 10.5281/zenodo.17704679
-
[29]
The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK
Verity Hill et al. “The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK”. In:Virus Evolution8.2 (2022), veac080.doi:10.1093/ve/veac080
-
[30]
John Huddleston et al. “Integrating genotypes and phenotypes improves long-term forecasts of seasonal in- fluenza A/H3N2 evolution”. In:eLife9 (2020), e60067.doi:10.7554/eLife.60067. 21
-
[31]
https://github.com/hubverse-org
The Consortium of Infectious Disease Modeling Hubs.The hubverse: open tools for collaborative modeling. https://github.com/hubverse-org. GitHub release v5.0.0, 17 Jan 2025, Accessed: 2025-06-13. 2025
2025
-
[32]
Real-Time Projections of SARS-CoV-2 B.1.1.7 Variant in a University Setting, Texas, USA
Kaitlyn E Johnson et al. “Real-Time Projections of SARS-CoV-2 B.1.1.7 Variant in a University Setting, Texas, USA”. In:Emerging Infectious Diseases27.12 (Dec. 2021), pp. 3188–3190.doi:10.3201/eid2712. 210652.url:https://doi.org/10.3201/eid2712.210652
-
[33]
Evaluating probabilistic forecasts with scoringRules
Alexander Jordan, Fabian Kr¨ uger, and Sebastian Lerch. “Evaluating probabilistic forecasts with scoringRules”. In:Journal of Statistical Software90 (2019), pp. 1–37
2019
-
[34]
SARS-CoV-2 vaccine strain selection: Guidance from influenza
Florian Krammer and Nadine Rouphael. “SARS-CoV-2 vaccine strain selection: Guidance from influenza”. In:Open Forum Infectious Diseases10.6 (2023), ofad239.doi:10.1093/ofid/ofad239
-
[35]
Sojung Lee, C´ ecile Viboud, and Trevor Bedford. “Reproducible and later vaccine strain selection can improve vaccine match to A/H3N2 seasonal influenza viruses”. In:npj Vaccines10 (2025), p. 23.doi:10.1038/s41541- 025-01292-w
-
[36]
Challenges of COVID-19 Case Forecasting in the US, 2020–2021
Velma K Lopez et al. “Challenges of COVID-19 Case Forecasting in the US, 2020–2021”. In:PLOS Compu- tational Biology20.5 (2024), e1011200.doi:10.1371/journal.pcbi.1011200
-
[37]
Dengue serotypes and epidemic dynamics in Brazil: a spatiotemporal perspective
Camila Lorenz, Philippe Lemey, and Simon Dellicour. “Dengue serotypes and epidemic dynamics in Brazil: a spatiotemporal perspective”. In:Travel Medicine and Infectious Disease68 (2025), p. 102948.doi:10.1016/ j.tmaid.2025.102948.url:https://doi.org/10.1016/j.tmaid.2025.102948
-
[38]
Sarabeth M. Mathis et al. “Evaluation of FluSight influenza forecasting in the 2021–22 and 2022–23 seasons with a new target laboratory-confirmed influenza hospitalizations”. In:Nature Communications15 (2024), p. 6289.doi:10.1038/s41467-024-50601-9
-
[39]
Evidence of latency reshapes our understanding of Ebola virus reservoir dynamics
John T McCrone et al. “Evidence of latency reshapes our understanding of Ebola virus reservoir dynamics”. In:bioRxiv(Oct. 2025). Preprint.doi:10.1101/2025.10.17.683141.url:https://www.biorxiv.org/ content/10.1101/2025.10.17.683141v1
work page doi:10.1101/2025.10.17.683141.url:https://www.biorxiv.org/ 2025
-
[40]
S. Molan et al. “Drivers of COVID-19 variant wave dynamics: inferring oncoming wave size using global data with genomics”. In:medRxiv(2025).doi:10.1101/2025.09.16.25335896. eprint:https://www.medrxiv. org/content/early/2025/09/18/2025.09.16.25335896.full.pdf.url:https://www.medrxiv.org/ content/early/2025/09/18/2025.09.16.25335896
-
[41]
Sang Woo Park et al. “Inferring the differences in incubation-period and generation-interval distributions of the Delta and Omicron variants of SARS-CoV-2”. In:Proceedings of the National Academy of Sciences120.22 (2023), e2221887120.doi:10.1073/pnas.2221887120. eprint:https://www.pnas.org/doi/pdf/10.1073/ pnas.2221887120.url:https://www.pnas.org/doi/abs/...
-
[42]
Open-source database for human viral pathogen genomic data
Pathoplexus.Pathoplexus. Open-source database for human viral pathogen genomic data. 2024.url:https: //pathoplexus.org
2024
-
[43]
Prabasaj Paul et al. “Genomic Surveillance for SARS-CoV-2 Variants Circulating in the United States, De- cember 2020–May 2021”. In:MMWR. Morbidity and Mortality Weekly Report70.23 (2021), pp. 846–850.doi: 10.15585/mmwr.mm7023a3
-
[44]
Abigail P. Paulos et al. “Detection of Hemagglutinin H5 Influenza A Virus RNA and Model of Potential Inputs in an Urban California Sewershed”. In:Environmental Science & Technology59.31 (2025), pp. 16168–16179. doi:10.1021/acs.est.4c14792
-
[45]
Considerable escape of SARS-CoV-2 Omicron to antibody neutralization
Delphine Planas et al. “Considerable escape of SARS-CoV-2 Omicron to antibody neutralization”. In:Nature 602.7898 (2022), pp. 671–675
2022
-
[46]
Generation of SARS-CoV-2 escape mutations by monoclonal antibody ther- apy
Manon Ragonnet-Cronin et al. “Generation of SARS-CoV-2 escape mutations by monoclonal antibody ther- apy”. In:Nature Communications14.1 (2023), p. 3334.doi:10.1038/s41467-023-37826-w.url:https: //doi.org/10.1038/s41467-023-37826-w. 22
-
[47]
A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology
Andrew Rambaut et al. “A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology”. In:Nature Microbiology(2020).doi:10.1038/s41564-020-0770-5
-
[48]
Evan L. Ray et al. “Comparing trained and untrained probabilistic ensemble forecasts of COVID-19 cases and deaths in the United States”. In:International Journal of Forecasting39 (2023), pp. 1366–1383.doi: 10.1016/j.ijforecast.2022.06.005
-
[49]
Nicholas G Reich et al.Collaborative hubs: making the most of predictive epidemic modeling. 2022
2022
-
[50]
Nicholas G. Reich et al. “A collaborative multiyear, multimodel assessment of seasonal influenza forecasting in the United States”. In:Proceedings of the National Academy of Sciences116.8 (2019), pp. 3146–3154.doi: 10.1073/pnas.1812594116
-
[51]
https://github.com/reichlab/variant- nowcast-hub
Reich Lab at UMass-Amherst.US SARS-CoV-2 Variant Nowcast Hub. https://github.com/reichlab/variant- nowcast-hub. Accessed: 2024-12-27. 2024
2024
-
[52]
Martina L. Reichmuth, Emma B. Hodcroft, and Christian L. Althaus. “Importation of Alpha and Delta variants during the SARS-CoV-2 epidemic in Switzerland: Phylogenetic analysis and intervention scenarios”. In:PLoS Pathogens19.8 (Aug. 2023), e1011553.doi:10.1371/journal.ppat.1011553
-
[53]
Thomas Robacker et al.Evaluating predictions of multi-class population proportions: A variant nowcasting case study.work in progress
-
[54]
https://next.nextstrain.org/nextclade/sars- cov-2
Cornelius Roemer and Richard Neher.SARS-CoV-2 phylogeny. https://next.nextstrain.org/nextclade/sars- cov-2. 2024
2024
-
[55]
GISAID: Global initiative on sharing all influenza data – from vision to reality
Yuelong Shu and John McCauley. “GISAID: Global initiative on sharing all influenza data – from vision to reality”. In:Eurosurveillance22.13 (2017).doi:10.2807/1560-7917.ES.2017.22.13.30494
-
[56]
Comparative analysis of Mpox clades: epidemiology, transmission dynamics, and detection strategies
Shriyansh Srivastava et al. “Comparative analysis of Mpox clades: epidemiology, transmission dynamics, and detection strategies”. In:BMC Infectious Diseases25 (Oct. 2025), p. 1290.doi:10 . 1186 / s12879 - 025 - 11784-8.url:https://pmc.ncbi.nlm.nih.gov/articles/PMC12516921/
2025
-
[57]
Leveraging global genomic sequencing data to estimate local variant dynamics
Zachary Susswein et al. “Leveraging global genomic sequencing data to estimate local variant dynamics”. In: medRxiv(2023).doi:10.1101/2023.01.02.23284123. eprint:https://www.medrxiv.org/content/early/ 2023/03/20/2023.01.02.23284123.full.pdf.url:https://www.medrxiv.org/content/early/2023/ 03/20/2023.01.02.23284123
-
[58]
Python Package Index
Becky Sweger et al.cladetime: Python interface for accessing Nextstrain SARS-CoV-2 sequence and clade data. Python Package Index. Available at https://pypi.org/project/cladetime/. 2024.url:https://github. com/reichlab/cladetime
2024
-
[59]
Founder effects arising from gathering dynamics systematically bias emerging pathogen surveillance
Bradford P Taylor and William P Hanage. “Founder effects arising from gathering dynamics systematically bias emerging pathogen surveillance”. In:eLife Sciences Publications, Ltd(Mar. 2025).doi:10.7554/elife. 104201.1.url:http://dx.doi.org/10.7554/eLife.104201.1
-
[60]
Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa
Houriiyah Tegally et al. “Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa”. In: Nature Medicine28.9 (Sept. 2022), pp. 1785–1790.doi:10.1038/s41591-022-01911-2
-
[61]
The evolution and biology of SARS-CoV-2 variants
Amalio Telenti, Emma B Hodcroft, and David L Robertson. “The evolution and biology of SARS-CoV-2 variants”. In:Cold Spring Harbor perspectives in medicine12.5 (2022), a041390
2022
-
[62]
Technical Briefing
UK Health Security Agency.SARS-CoV-2 variants of concern and variants under investigation in Eng- land: Technical briefing 38. Technical Briefing. Published: 11 March 2022. Mar. 2022.url:https : / / assets . publishing . service . gov . uk / media / 622b4a20e90e070ed8233a05 / Technical - Briefing - 38 - 11March2022.pdf
2022
-
[63]
Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England
Erik Volz et al. “Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England”. In:Nature593 (2021), pp. 266–269.doi:10.1038/s41586-021-03470-x.url:https://www.nature.com/articles/s41586-021- 03470-x. 23
work page doi:10.1038/s41586-021-03470-x.url:https://www.nature.com/articles/s41586-021- 2021
-
[64]
Debra A. Wadford et al. “Implementation of California COVIDNet – a multi-sector collaboration for statewide SARS-CoV-2 genomic surveillance”. In:Frontiers in Public Health11 (2023), p. 1249614.doi:10 . 3389 / fpubh.2023.1249614
arXiv 2023
-
[65]
Collaborative nowcasting of COVID-19 hospitalization incidences in Germany
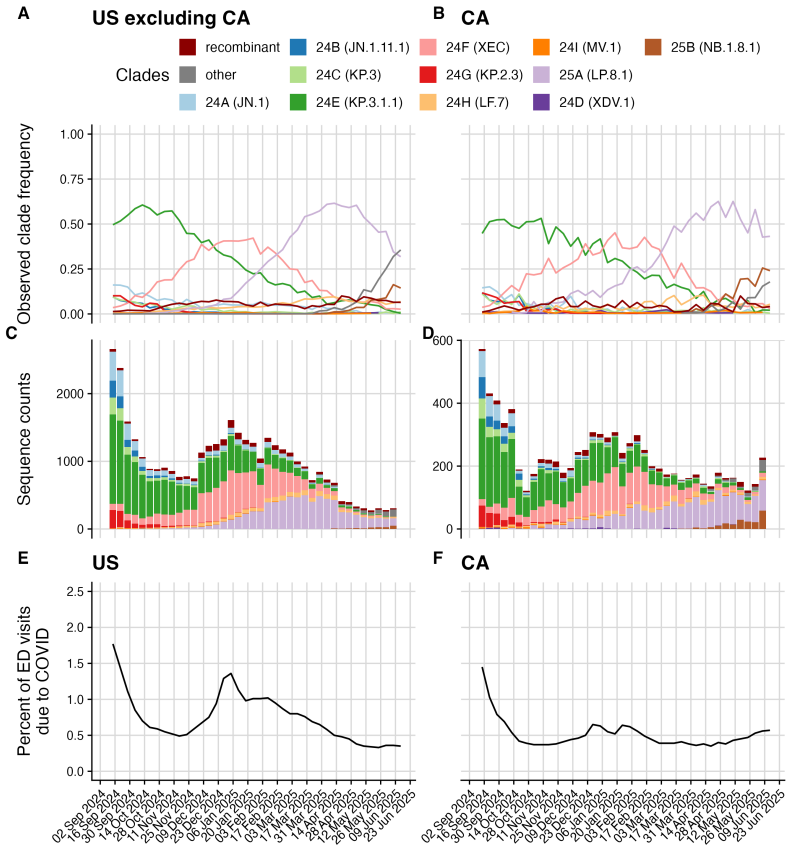
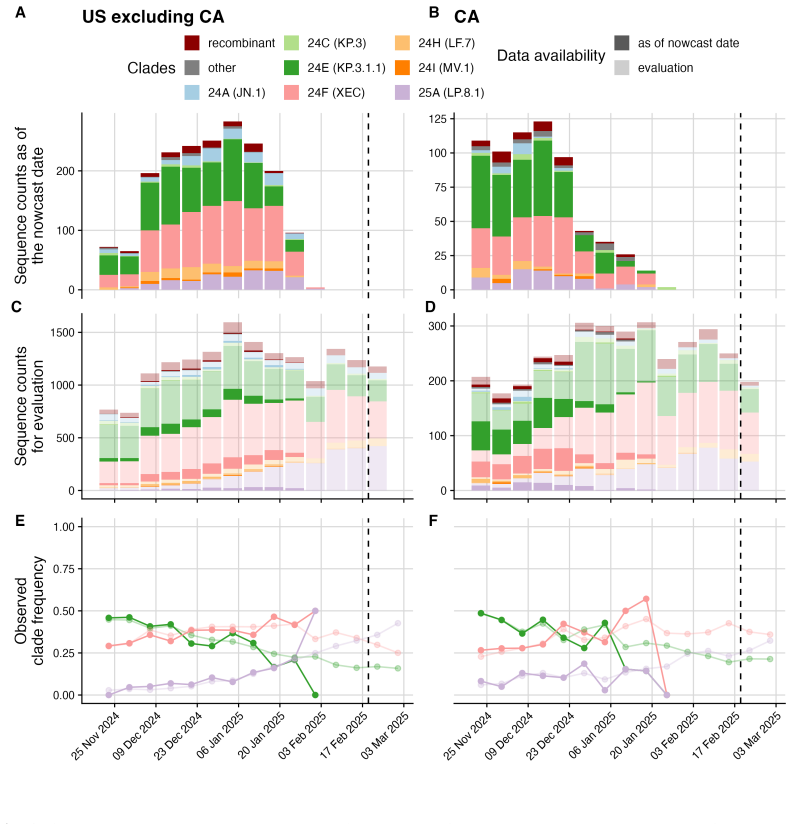
Daniel Wolffram et al. “Collaborative nowcasting of COVID-19 hospitalization incidences in Germany”. In: PLOS Computational Biology19.8 (2023), e1011394.doi:10.1371/journal.pcbi.1011394. 24 Figure 2: Weekly SARS-CoV-2 variant dynamics in the U.S. from September 2nd, 2024 to June 14th, 2025 using the final set of sequences available 90 days after the last ...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.