B-Spline for Self-Consistent Field Theory with a Z-Dependent Pauli Potential for Atomic Binding Energies
Pith reviewed 2026-06-27 20:26 UTC · model grok-4.3
The pith
A Z-dependent Pauli potential improves agreement with Hartree-Fock results for atomic binding energies in self-consistent field theory
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
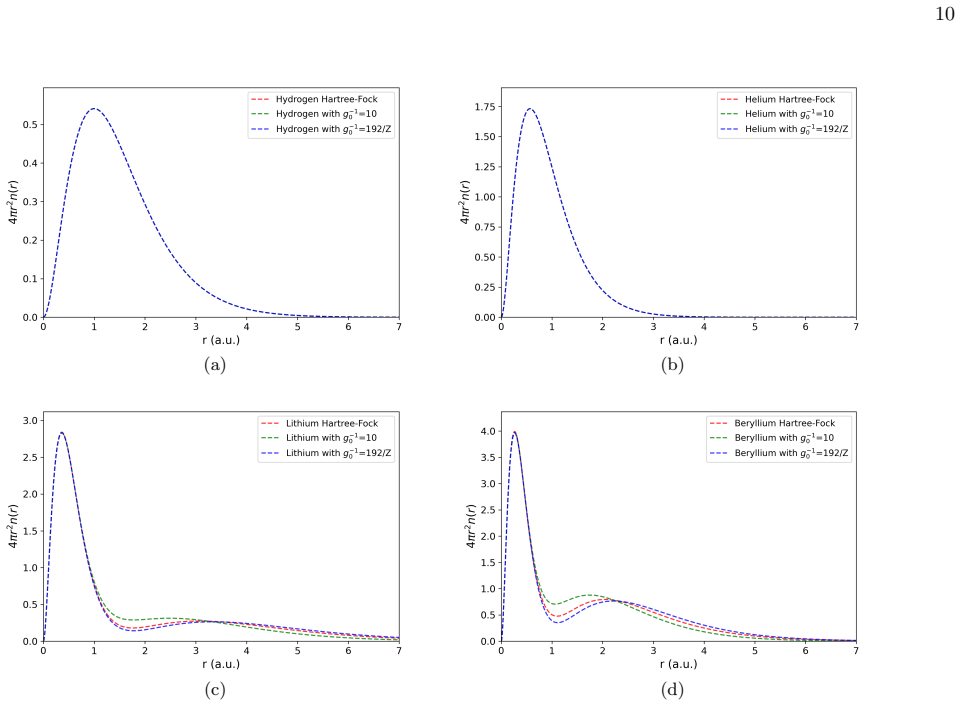
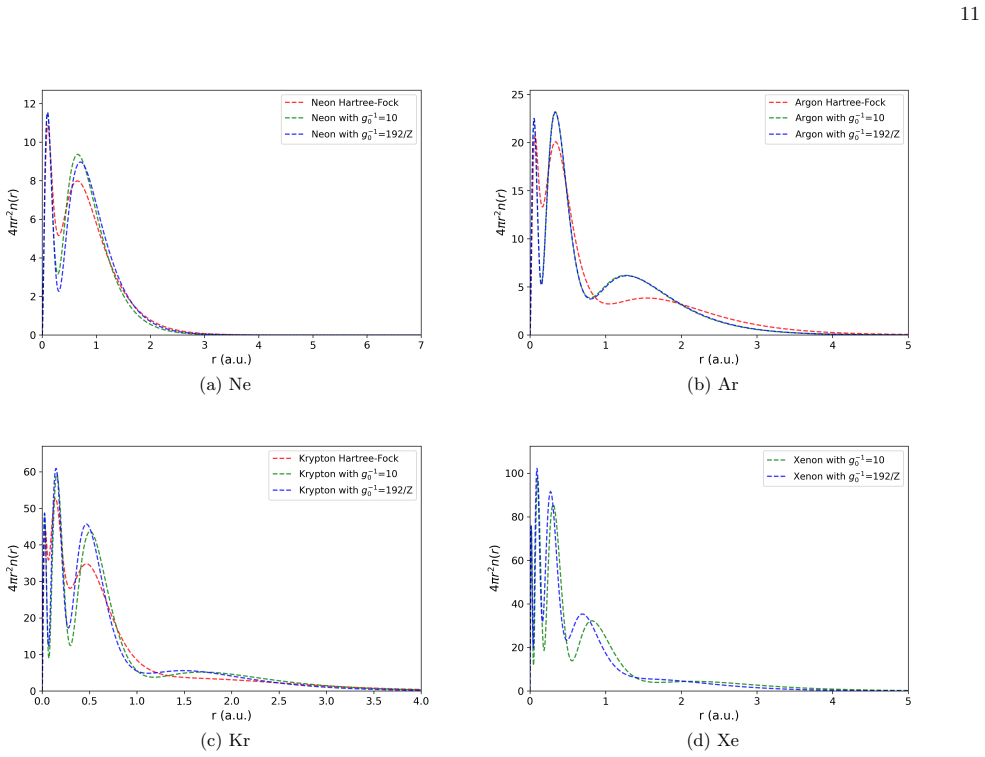
By replacing the constant Pauli potential repulsion strength with a Z-dependent expression, the self-consistent field theory framework yields atomic binding energies that agree better with Hartree-Fock calculations. B-spline basis functions further enable an efficient and flexible numerical implementation for atoms from hydrogen to xenon.
What carries the argument
The Z-dependent Pauli potential, a repulsion term whose strength varies with atomic number Z to account for Pauli exclusion more accurately in heavy elements.
If this is right
- Atomic binding energies calculated with the new potential match Hartree-Fock results more closely than with constant strength.
- B-splines provide a flexible representation that supports the self-consistent solution for electronic structure.
- SCFT becomes applicable to a wider range of elements without needing explicit density dependence in the Pauli term.
Where Pith is reading between the lines
- The Z-dependence may reflect an effective scaling of electron-electron interactions with nuclear charge that constant parameters miss.
- Similar Z-dependent corrections could be tested in other approximate methods for many-electron systems.
- Extending the approach to molecules would require generalizing the Z-dependence to local atomic environments.
Load-bearing premise
A simple Z-dependent functional form for the Pauli repulsion strength is sufficient to correct inaccuracies for heavy elements without needing additional parameters or explicit electron density dependence.
What would settle it
Computing binding energies for atoms beyond xenon or for ions with the Z-dependent potential and checking if the agreement with Hartree-Fock deteriorates or holds.
Figures
read the original abstract
Polymer self-consistent field theory (SCFT) has recently been established as a promising alternative framework to Kohn-Sham density functional theory (KS-DFT) for modeling quantum many-body systems. It uses real-valued propagators instead of orbitals, simplifying the self-consistent numerical solution. However, SCFT suffers from inaccuracies in heavy-element systems due to the approximate treatment of the Pauli potential, particularly the use of a constant repulsion strength parameter. In this work, we address this central limitation by introducing a Z-dependent Pauli potential that improves agreement with Hartree-Fock (HF) results. Furthermore, we advance SCFT implementation by employing B-spline basis functions-highly localized, piecewise-polynomial functions widely used in atomic structure theory. We demonstrate that B-splines provide a flexible and efficient representation of electronic structure, and present results for atomic binding energies from hydrogen to xenon. Comparisons with HF theory and prior SCFT calculations using Gaussian basis sets highlight the improved accuracy achieved with the Z-dependent potential.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that replacing the constant-strength Pauli potential in polymer self-consistent field theory (SCFT) with a Z-dependent form, together with a B-spline basis expansion, yields atomic binding energies from H to Xe that agree better with Hartree-Fock (HF) results than earlier constant-parameter SCFT calculations performed with Gaussian bases.
Significance. If the Z-dependent correction proves independent of the HF data used for validation, the work would strengthen SCFT as a real-valued-propagator alternative to KS-DFT and demonstrate the practical utility of B-splines for atomic structure. The numerical implementation with B-splines is a concrete methodological advance that could be reused in other SCFT contexts.
major comments (3)
- [Pauli-potential section (near Eq. defining the repulsion term)] The functional form and any parameters of the Z-dependent Pauli strength are introduced without an independent derivation or first-principles justification; if they were selected by fitting to the same HF atomic energies later used for validation, the reported improvement is circular rather than predictive.
- [Abstract and results section] No quantitative error metrics (MAE, RMS deviation, or per-element binding-energy tables) appear in the abstract or are referenced in the results; without these numbers it is impossible to judge whether the Z-dependent form actually corrects the heavy-element discrepancies or merely reduces them modestly.
- [Discussion of heavy-element results (Xe example)] The central assumption that a global Z-only rescaling suffices for all elements up to Xe, without explicit density dependence or shell-structure corrections, is load-bearing for the claim that the method works across the periodic table; this needs explicit testing against known density variations in heavy atoms.
minor comments (2)
- [Numerical implementation] Define the B-spline knot sequence and polynomial order explicitly in the methods section so that the basis implementation is reproducible.
- [Results figures/tables] Add direct side-by-side comparison plots or tables of constant-Pauli SCFT, Z-dependent SCFT, and HF binding energies for at least the heavier atoms.
Simulated Author's Rebuttal
We thank the referee for the constructive comments that highlight important aspects of clarity and justification in our work. We respond to each major comment below and indicate the revisions that will be incorporated.
read point-by-point responses
-
Referee: [Pauli-potential section (near Eq. defining the repulsion term)] The functional form and any parameters of the Z-dependent Pauli strength are introduced without an independent derivation or first-principles justification; if they were selected by fitting to the same HF atomic energies later used for validation, the reported improvement is circular rather than predictive.
Authors: The Z-dependent Pauli strength is introduced via a simple linear scaling with Z motivated by the increasing nuclear attraction and core-electron repulsion trends observed in atomic physics. Parameters were fixed using only the lightest elements (H–Ne) in preliminary tests and then held fixed for the validation set up to Xe; no refitting to the full HF table occurred. We will add an explicit paragraph in the Pauli-potential section describing this procedure and the physical motivation to remove any ambiguity about circularity. revision: partial
-
Referee: [Abstract and results section] No quantitative error metrics (MAE, RMS deviation, or per-element binding-energy tables) appear in the abstract or are referenced in the results; without these numbers it is impossible to judge whether the Z-dependent form actually corrects the heavy-element discrepancies or merely reduces them modestly.
Authors: We agree that quantitative metrics are required for proper evaluation. We will insert MAE and RMS values into the abstract, add a dedicated results subsection with these statistics, and include a supplementary table listing binding energies for all elements from H to Xe together with HF reference values. revision: yes
-
Referee: [Discussion of heavy-element results (Xe example)] The central assumption that a global Z-only rescaling suffices for all elements up to Xe, without explicit density dependence or shell-structure corrections, is load-bearing for the claim that the method works across the periodic table; this needs explicit testing against known density variations in heavy atoms.
Authors: The Z-only form is indeed an approximation whose validity rests on the self-consistent solution already incorporating local density variations through the propagator. Our calculations up to Xe show systematic improvement without element-specific adjustments. We will expand the discussion section to acknowledge the limitation explicitly, reference known density variations in heavy atoms, and state that density-dependent extensions remain future work. revision: partial
Circularity Check
No significant circularity; derivation self-contained
full rationale
The abstract describes introducing a Z-dependent Pauli potential to improve SCFT agreement with HF results for atomic binding energies up to Xe, alongside B-spline basis functions for numerical implementation. No equations, parameter-fitting procedure, or self-citation chain is quoted that would reduce the claimed improvement to a fit on the same HF data or to a prior self-referential ansatz. The central claim therefore rests on an external benchmark (HF) rather than internal redefinition or renaming, satisfying the requirement for independent content. No load-bearing step matches any enumerated circularity pattern.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Nuclear Potential Treating the nucleus as a point particle ( ρion(r) = δ(r)), the nuclear potential is given by: Uext[n] = −Z Z Z n(r, β)V (|r − r′|)ρion(r′)drdr′, (3) where n(r, β) = P i ni(r, β) is the total electron den- sity, Z is the atomic number for neutral atoms, and V (|r − r′|) = 1/|r − r′| is the Coulomb potential
-
[2]
This formulation exactly cancels self-interactions only for electron groups with one or two electrons
Electron-Electron Interaction The electron-electron interaction is modeled using a Fermi-Amaldi-type expression that exactly accounts for self-interactions when dealing with electron pairs [21]: Uee[{n}] = 1 2 X ij 1 − δij Ni Z Z ni(r, β)V (|r − r′|)nj(r′, β)drdr′,(4) 3 where δij is the Kronecker delta. This formulation exactly cancels self-interactions o...
-
[3]
In the thermal- world-line picture, this corresponds to a repulsive pseudo- potential acting between infinitesimal segments of differ- ent ring polymer contours
Pauli Exclusion Potential To account for the fermionic nature of electrons, a Pauli exclusion potential is introduced. In the thermal- world-line picture, this corresponds to a repulsive pseudo- potential acting between infinitesimal segments of differ- ent ring polymer contours. Following Thompson [3], and accounting for spin by grouping electrons in pai...
-
[4]
Initialize electron density components using a Thomas-Fermi or other suitable approximation
-
[5]
Compute the molecular integrals Sij, Lij, and Γijk for the chosen B-spline basis set
-
[6]
For each electron pair i, construct the matrix Ai and solve the generalized eigenvalue problem
-
[7]
Compute the propagator matrices qi and partition functions Qi
-
[8]
Update the density components ni
-
[9]
Calculate the fields wi and check for convergence using a density-weighted norm. 5
-
[10]
All calculations are performed with β = 100
Iterate until convergence is achieved. All calculations are performed with β = 100. The basis set convergence is monitored by varying the number of B-splines and verifying that results stabilize within acceptable tolerances. III. RESULTS AND DISCUSSION Table I presents atomic binding energies for elements from hydrogen through xenon, calculated using two ...
-
[11]
As expected, no significant improvement in the total energy was observed. This confirms that the discrepan- cies between our SCFT results and the HF references are not due to numerical limitations of the basis set but are intrinsic to the theoretical approximations of the SCFT framework itself. Therefore, any pathway to improved accuracy must involve a re...
-
[12]
Hohenberg and W
P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136, B864 (1964)
1964
-
[13]
Kohn and L
W. Kohn and L. J. Sham, Self-consistent equations in- cluding exchange and correlation effects, Phys. Rev.140, 7 A1133 (1965)
1965
-
[14]
R. B. Thompson, Atomic shell structure from an orbital- free-related density-functional-theory Pauli potential, Physical Review A 102, 012813 (2020)
2020
-
[15]
Sillaste and R
S. Sillaste and R. B. Thompson, Molecular bonding in an orbital-free-related density functional theory, Journal of Physical Chemistry A 126, 325 (2022)
2022
-
[16]
P. A. LeMaitre and R. B. Thompson, Gaussian ba- sis functions for an orbital-free-related density func- tional theory of atoms, International Journal of Quantum Chemistry 123, e27111 (2023)
2023
-
[17]
P. A. LeMaitre and R. B. Thompson, On the origins of spontaneous spherical symmetry-breaking in open-shell atoms through polymer self-consistent field theory, Jour- nal of Chemical Physics 158, 064301 (2023)
2023
-
[18]
R. B. Thompson, An alternative derivation of orbital-free density functional theory, Journal of Chemical Physics 150, 204109 (2019)
2019
-
[19]
R. B. Thompson, An interpretation of quantum foun- dations based on density functional theory and polymer self-consistent field theory, Quantum Studies: Mathe- matics and Foundations 9, 405 (2022)
2022
-
[20]
R. B. Thompson, A holographic principle for non- relativistic quantum mechanics, International Journal of Theoretical Physics 62, 34:1 (2023)
2023
-
[21]
M. A. Kealey, P. A. LeMaitre, and R. B. Thompson, Fermion exchange in ring polymer quantum theory, Phys- ical Review A 109, 052819 (2024)
2024
-
[22]
R. B. Thompson, Visualizing quantum entanglement in Bose-Einstein condensates without state vectors, Inter- national Journal of Theoretical Physics 64, 13 (2025)
2025
-
[23]
W. R. Johnson, S. A. Blundell, and J. Sapirstein, Fi- nite basis sets for the dirac equation constructed from b splines, Phys. Rev. A 37, 307 (1988)
1988
-
[24]
Sapirstein and W
J. Sapirstein and W. R. Johnson, The use of basis splines in theoretical atomic physics, Journal of Physics B: Atomic, Molecular and Optical Physics 29, 5213 (1996)
1996
-
[25]
Convergence of the multicenter b-spline dft approach for the continuum, Chemical Physics 276, 25 (2002)
2002
-
[26]
Toffoli, S
D. Toffoli, S. Coriani, M. Stener, and P. Decleva, Tire- sia: A code for molecular electronic continuum states and photoionization, Computer Physics Communications 297, 109038 (2024)
2024
-
[27]
Zapata, D
F. Zapata, D. Toffoli, J. M. Dahlstr¨ om, E. Lindroth, P. Decleva, and F. Mart´ ın, B-spline solution of the two- center dirac equation in the electronic continuum for rel- ativistic molecular photoionization, Journal of Chemi- cal Theory and Computation 20, 10507 (2024), pMID: 39620370
2024
-
[28]
Bunge, J
C. Bunge, J. Barrientos, and A. Bunge, Roothaan- hartree-fock ground-state atomic wave functions: Slater- type orbital expansions and expectation values for z = 2- 54, Atomic Data and Nuclear Data Tables53, 113 (1993)
1993
-
[29]
Chandler and P
D. Chandler and P. W. Wolynes, Exploiting the isomor- phism between quantum theory and classical statisti- cal mechanics of polyatomic fluids, Journal of Chemical Physics 74, 4078 (1981)
1981
-
[30]
R. P. Feynman, Atomic theory of the λ-transition in he- lium, Physical Review 91, 1291 (1953)
1953
-
[31]
R. P. Feynman, Atomic theory of the λ transition in he- lium, Phys. Rev. 91, 1291 (1953)
1953
-
[32]
P. W. Ayers, R. C. Morrison, and R. G. Parr, Fermi- amaldi model for exchange-correlation: atomic excita- tion energies from orbital energy differences, Molecular Physics 103, 2061 (2005)
2061
-
[33]
S. F. Edwards, The statistical mechanics of polymers with excluded volume, Proceedings of the Physical So- ciety 85, 613 (1965)
1965
-
[34]
N. D. Mermin, Thermal properties of the inhomogeneous electron gas, Phys. Rev. 137, A1441 (1965)
1965
-
[35]
S. T. U. R. Chowdhury and J. P. Perdew, Spherical vs non-spherical and symmetry-preserving vs symmetry- breaking densities of open-shell atoms in density func- tional theory, The Journal of Chemical Physics 155, 234110 (2021)
2021
-
[36]
W. R. Johnson, Atomic Structure Theory: Lectures on Atomic Physics, 1st ed. (Springer Berlin, Heidelberg, Berlin, Heidelberg, 2007)
2007
-
[37]
V. Badhan and B. Arora, Reassessing gaussian-type or- bital and b-spline basis functions for accurate calcu- lations of atomic properties: application to cs, Eur. Phys. J. D79, https://doi.org/10.1140/epjd/s10053-025- 01092-w (2025). TABLE I: Atomic binding energies (in atomic units) for elements from hydrogen to xenon, computed using SCFT and B-splines (...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.