scCBGM: Interpretable Single-Cell Counterfactual Editing
Pith reviewed 2026-06-27 22:11 UTC · model grok-4.3
The pith
scCBGM adapts concept bottleneck models with skip connections and a cross-covariance penalty to enable interpretable counterfactual editing of single cells.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
scCBGM is a framework that adapts concept bottleneck generative models to single-cell RNA sequencing data through decoder skip connections and a cross-covariance penalty for disentanglement, then extends the approach to flow matching models, thereby supporting precise concept-guided counterfactual editing in both encoding-decoding and generation regimes while outperforming baselines on combinatorial generalization tasks.
What carries the argument
The concept bottleneck architecture adapted via decoder skip connections and a cross-covariance penalty that promotes disentanglement in the learned concept representations.
If this is right
- Concept-guided editing becomes possible in both reconstruction and unconditional generation regimes through the flow-matching extension.
- Combinatorial generalization improves across multiple real single-cell datasets compared with prior methods.
- Cell-level validation on synthetic data with known counterfactuals becomes feasible alongside population-level benchmarks.
- Disentangled concepts allow separate control of distinct biological factors during editing without dimensional restrictions.
Where Pith is reading between the lines
- The approach could reduce reliance on exhaustive wet-lab perturbation screens by simulating responses to untested condition combinations.
- Disentangled concepts might map onto known biological pathways, letting users inspect which factors drive a given edit.
- Scaling the method to multimodal data such as paired RNA and protein measurements would test whether the same skip-connection and penalty design transfers.
- If the cross-covariance term remains effective at larger concept counts, the framework could support finer-grained editing than dimension-constrained alternatives.
Load-bearing premise
The synthetic benchmark with ground-truth counterfactuals accurately reflects the complexities and noise of real single-cell data, and the cross-covariance penalty effectively promotes disentanglement without introducing new constraints.
What would settle it
An independent set of real perturbation experiments where the cell-level counterfactual predictions generated by scCBGM systematically diverge from the measured post-perturbation expression profiles.
Figures



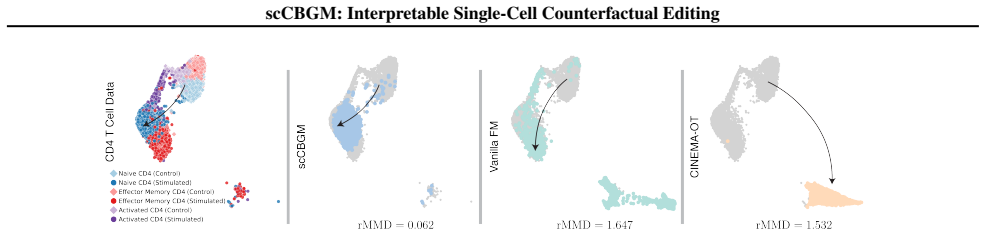








read the original abstract
Understanding cellular phenotypes and how they respond to perturbations is critical for disease biology and therapeutic design. Single-cell RNA sequencing enables characterization at cellular resolution, yet the combinatorial space of conditions makes exhaustive experimental mapping infeasible. We introduce single-cell Concept Bottleneck Generative Models (scCBGM), a framework for interpretable and precise counterfactual editing of individual cells. scCBGM adapts concept bottleneck architectures for single-cell data through decoder skip connections and a cross-covariance penalty that promotes disentanglement without dimensional constraints. We extend the framework to flow matching models, enabling concept-guided editing in both encoding-decoding and generation regimes. To enable rigorous evaluation, we develop a synthetic benchmark with ground-truth counterfactuals. Across multiple real datasets, scCBGM demonstrates superior performance in combinatorial generalization and counterfactual prediction, supported by cell-level validation on synthetic data and population-level benchmarks on real datasets.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces single-cell Concept Bottleneck Generative Models (scCBGM) that adapt concept bottleneck architectures to single-cell RNA-seq via decoder skip connections and a cross-covariance penalty for disentanglement (without dimensional constraints), extends the approach to flow matching models for concept-guided editing, constructs a synthetic benchmark with ground-truth counterfactuals, and reports superior performance in combinatorial generalization and counterfactual prediction on multiple real datasets via cell-level synthetic validation and population-level real-data benchmarks.
Significance. If the central claims hold after addressing benchmark realism, the work would provide a useful interpretable framework for counterfactual editing in single-cell data, combining concept bottlenecks with generative modeling to support perturbation analysis in disease biology; the explicit synthetic benchmark with ground truth is a positive step toward rigorous evaluation in this domain.
major comments (2)
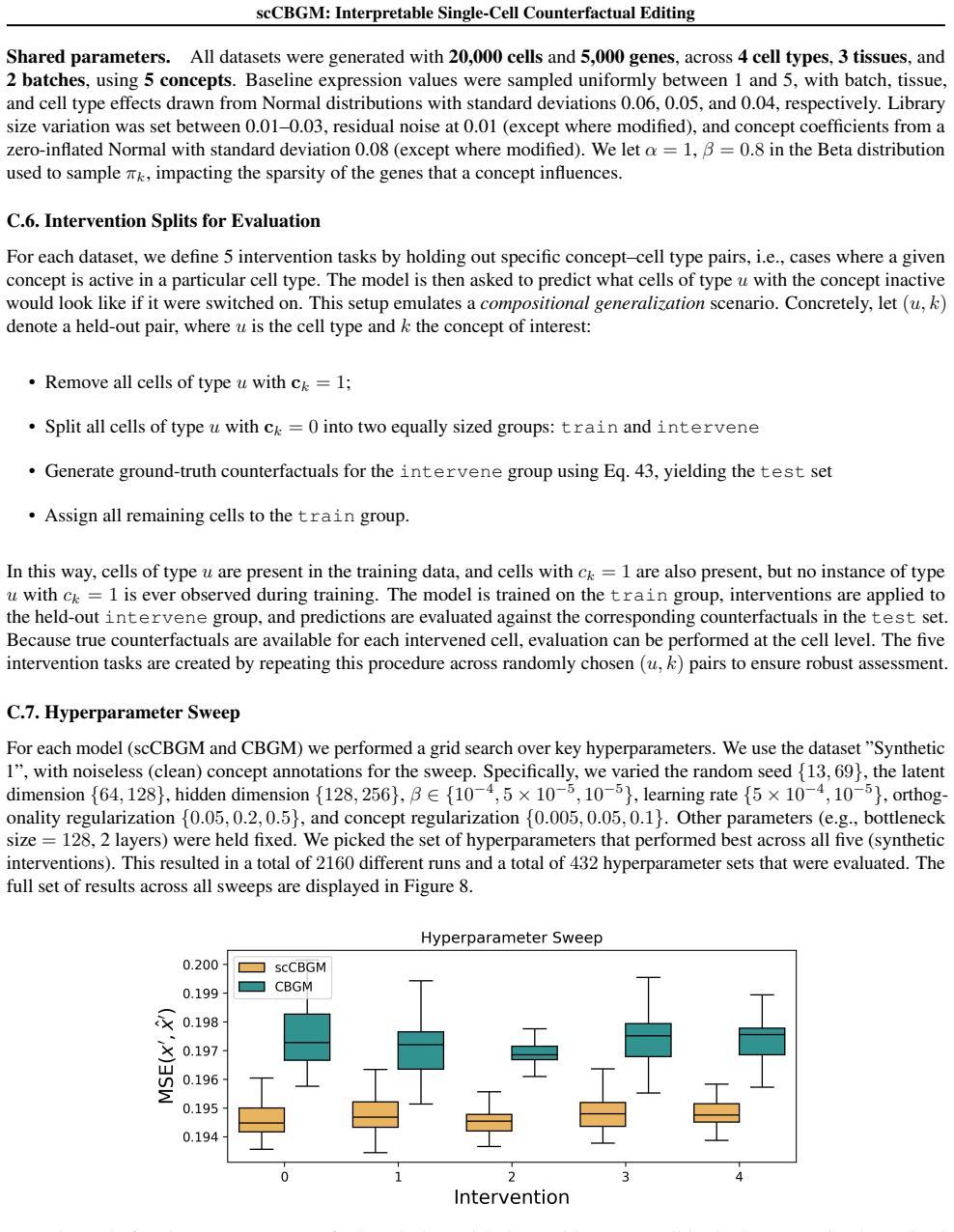
- [§4] §4 (Synthetic Benchmark): the generative process used to create the synthetic data with ground-truth counterfactuals is not shown to incorporate zero-inflation, high dropout rates, or batch effects typical of real scRNA-seq; without this, cell-level validation on synthetic data does not license the extrapolation that the same architecture and cross-covariance penalty will yield accurate edits on real data.
- [§5] §5 (Real-data Experiments): population-level metrics on real datasets cannot substitute for per-cell ground truth, so the combinatorial generalization claim rests entirely on the synthetic regime; the cross-covariance penalty's disentanglement effect is only demonstrated under the synthetic noise model and remains untested under realistic dropout and sparsity.
minor comments (2)
- The abstract asserts 'superior performance' without naming the exact baselines, metrics, or statistical tests; these details should be summarized in the abstract for clarity.
- Notation for the cross-covariance penalty term should be defined explicitly with an equation number in the methods section to allow direct comparison with related disentanglement penalties.
Simulated Author's Rebuttal
We thank the referee for their insightful comments on the synthetic benchmark and real-data evaluation. We address each major comment below, providing clarifications and indicating planned revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [§4] §4 (Synthetic Benchmark): the generative process used to create the synthetic data with ground-truth counterfactuals is not shown to incorporate zero-inflation, high dropout rates, or batch effects typical of real scRNA-seq; without this, cell-level validation on synthetic data does not license the extrapolation that the same architecture and cross-covariance penalty will yield accurate edits on real data.
Authors: We agree that our synthetic data generation process does not explicitly incorporate zero-inflation, high dropout rates, or batch effects characteristic of real scRNA-seq data. The primary goal of the synthetic benchmark is to provide ground-truth counterfactuals for evaluating combinatorial generalization at the cell level, a capability not available in real datasets. This allows us to rigorously test the model's ability to perform precise edits under controlled conditions. While we acknowledge this simplification limits direct claims about robustness to real noise distributions, the consistent superior performance observed on multiple real datasets using population-level metrics provides supporting evidence of practical utility. In the revised manuscript, we will expand the discussion of the synthetic benchmark to explicitly state its assumptions and limitations, and suggest future work on more realistic noise models. This constitutes a partial revision. revision: partial
-
Referee: [§5] §5 (Real-data Experiments): population-level metrics on real datasets cannot substitute for per-cell ground truth, so the combinatorial generalization claim rests entirely on the synthetic regime; the cross-covariance penalty's disentanglement effect is only demonstrated under the synthetic noise model and remains untested under realistic dropout and sparsity.
Authors: We concur that population-level metrics on real data cannot replace per-cell ground truth, and thus the strongest evidence for combinatorial generalization comes from the synthetic benchmark. On real datasets, our evaluation follows standard practices in the field by using population-level benchmarks for counterfactual prediction tasks. For the cross-covariance penalty, its role in promoting disentanglement is indeed illustrated and quantified in the synthetic setting where the underlying factors are known. On real data, we demonstrate its benefit through improved editing performance rather than direct disentanglement metrics. In revision, we will clarify the scope of these claims in the text and add a limitations paragraph noting that disentanglement under realistic noise remains to be further investigated. This is a partial revision. revision: partial
Circularity Check
No circularity in claimed derivation or predictions
full rationale
The paper presents an empirical framework evaluated on external synthetic and real datasets, with performance claims resting on benchmark comparisons rather than any derivation that reduces to its own fitted parameters or self-citations by construction. The abstract and context describe architectural adaptations, a penalty term, and a new benchmark, but contain no load-bearing steps where a 'prediction' or result is definitionally equivalent to an input (e.g., no fitted quantities renamed as predictions, no uniqueness theorems imported from self-citations, and no ansatzes smuggled via prior work). The central claims are supported by independent validation data, making the derivation self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
K., Gautam, D., Bevilacqua, B., Imran, A., Shah, R., Naghipourfar, M., Teyssier, N., Ilango, R., Nagaraj, S., Dong, M., et al
Adduri, A. K., Gautam, D., Bevilacqua, B., Imran, A., Shah, R., Naghipourfar, M., Teyssier, N., Ilango, R., Nagaraj, S., Dong, M., et al. Predicting cellular responses to perturbation across diverse contexts with state.BioRxiv, pp. 2025–06,
2025
-
[2]
arXiv preprint arXiv:2304.06129 (2023) 2, 4
Oikarinen, T., Das, S., Nguyen, L. M., and Weng, T.-W. Label-free concept bottleneck models.arXiv preprint arXiv:2304.06129,
-
[3]
Semantic image inversion and editing us- ing rectified stochastic differential equations,
Rout, L., Chen, Y ., Ruiz, N., Caramanis, C., Shakkottai, S., and Chu, W.-S. Semantic image inversion and editing using rectified stochastic differential equations.arXiv preprint arXiv:2410.10792,
-
[4]
Sanchez, P. and Tsaftaris, S. A. Diffusion causal models for counterfactual estimation.arXiv preprint arXiv:2202.10166,
-
[5]
Tam- ing rectified flow for inversion and editing.arXiv preprint arXiv:2411.04746, 2024
Wang, G., Liu, T., Zhao, J., Cheng, Y ., and Zhao, H. Mod- eling and predicting single-cell multi-gene perturbation responses with scLAMBDA.bioRxiv, 2024a. Wang, J., Pu, J., Qi, Z., Guo, J., Ma, Y ., Huang, N., Chen, Y ., Li, X., and Shan, Y . Taming rectified flow for inversion and editing.arXiv preprint arXiv:2411.04746, 2024b. Wenteler, A., Occhetta, M...
-
[6]
Xia, T., Ribeiro, F. D. S., Rasal, R. R., Kori, A., Mehta, R., and Glocker, B. Decoupled classifier-free guid- ance for counterfactual diffusion models.arXiv preprint arXiv:2506.14399,
work page internal anchor Pith review Pith/arXiv arXiv
-
[7]
Additional details on Singe-Cell Concept Bottleneck Generative Models A.1
12 scCBGM: Interpretable Single-Cell Counterfactual Editing Appendix A. Additional details on Singe-Cell Concept Bottleneck Generative Models A.1. A very short primer on counterfactuals We posit a structural causal model (SCM) M= (G, F, P(U)) with a directed acyclic graph G, endogenous variables V , exogenous noiseU, and assignmentsF={v i ←f i(pai, ui)}. ...
2000
-
[8]
We excluded megakaryocytes due to their low cell count (210 cells)
dataset comprises 24,264 cells across 8 broad-cell types, observed under two conditions: with and without IFN-β stimulation. We excluded megakaryocytes due to their low cell count (210 cells). Data was preprocessed usingscanpy(Wolf et al., 2018), involving median library size normalization, log-transformation of all counts, and filtering to the top 3000 m...
2018
-
[9]
PBMC dataset. To benchmark our model on high-fidelity edits that preserve cell phenotype while changing experimental conditions, we focused on identifying granular phenotypes consistent across stimulated and unstimulated cells. We first integrated the two conditions into a unified latent space using Harmony (Korsunsky et al., 2019). Within this unified la...
2019
-
[10]
For the 22 scCBGM: Interpretable Single-Cell Counterfactual Editing Kang et al. (2017) experiments, concepts were defined using these original broad-cell types, while stimulation predictions and held-out validations were conducted at the more granular subtype level. We ran our experiments over the same 4 random seeds for all models. Table 6 presents the h...
2017
-
[11]
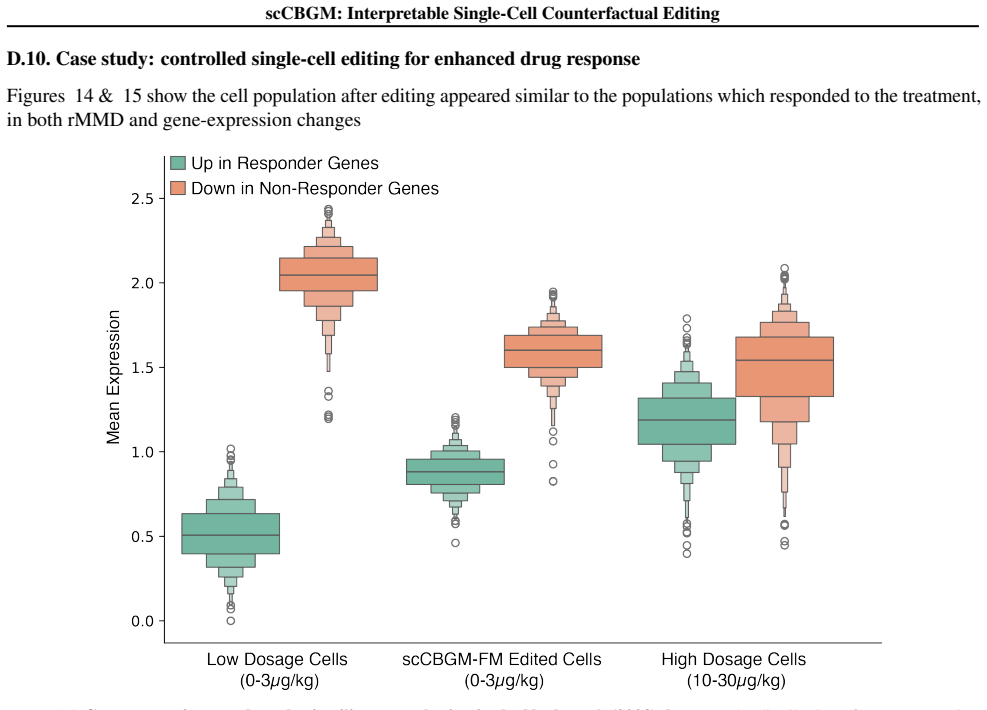
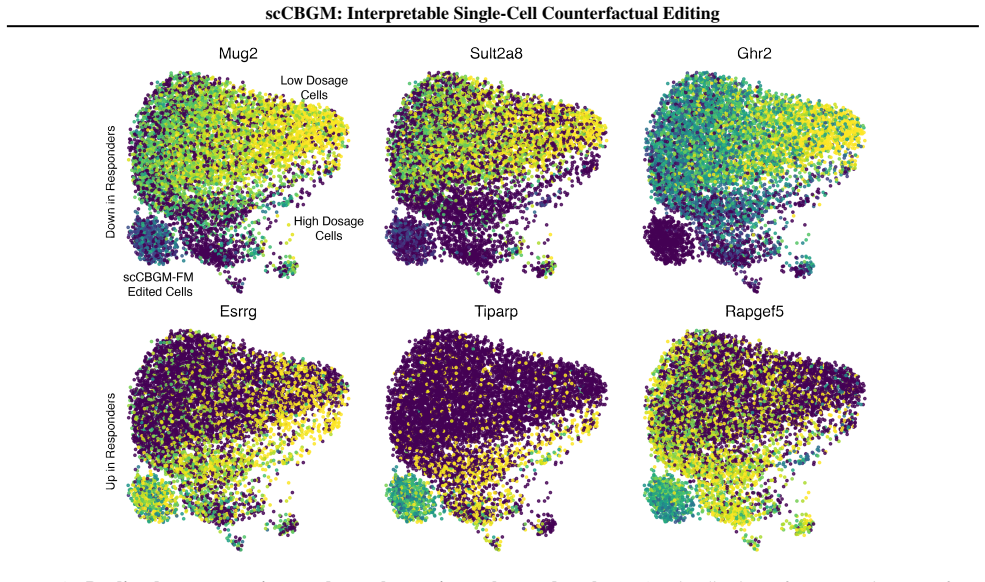
By comparing responder hepatic stellate cells (dosed at 10 and 30µg/kg) with non-responders (all other dosages), we identified differentially regulated pathways
using thedecouplerpackage (Badia-i Mompel et al., 2022). By comparing responder hepatic stellate cells (dosed at 10 and 30µg/kg) with non-responders (all other dosages), we identified differentially regulated pathways. Specifically, responder stellate cells exhibited high activity 24 scCBGM: Interpretable Single-Cell Counterfactual Editing in TGFβ, PI3K, ...
2022
-
[12]
Values are MSE (mean ± std), averaged over interventions (5) and seeds (4). 30 scCBGM: Interpretable Single-Cell Counterfactual Editing This trend is consistent with prior observations in the literature (Ismail et al., 2025), indicating that the model remains stable and effective even as the concept space expands. concepts MSE 5 0.19617±0.00195 20 0.19597...
2025
-
[13]
This stark reduction confirms that the known categorical variation is effectively isolated and decoupled 32 scCBGM: Interpretable Single-Cell Counterfactual Editing from the residual unknown layer. Fold 1 Fold 2 Fold 3 Fold 4 Fold 5 Mean±Std Stim in Known 0.572 0.577 0.555 0.545 0.541 0.5580±0.0160 Stim in Unknown 0.998 1.000 0.998 0.999 0.999 0.9988±0.00...
1989
-
[14]
Using a paired t-test over all 1479 configurations, scCBGM-FM was found to significantly outperform CV AE-FM across the full intervention space (p-val<1e −10) for all metrics. D.9.3. COMPLETENAULT ET AL. (2023)RESULTS In Tables 21, 22, and 23 we report the results of our experiment on the Nault et al. (2023) dataset on all available cell types for the rMM...
-
[15]
The successful recovery of the remaining 80% suggests that the model captures downstream regulatory effects beyond the direct inputs
We note that only 40 of these marker genes overlap with the total set of top 100 genes defining the manipulated pathway concepts (500 total). The successful recovery of the remaining 80% suggests that the model captures downstream regulatory effects beyond the direct inputs. D.11. Cell subtype accuracy To complement our benchmark, we evaluate whether edit...
2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.