On the Covalent Fields of Molecule-Surface Interactions
Pith reviewed 2026-06-27 17:54 UTC · model grok-4.3
The pith
Representing chemical affinity as a continuous covalent field resolves active site ambiguity and scaling relation issues.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
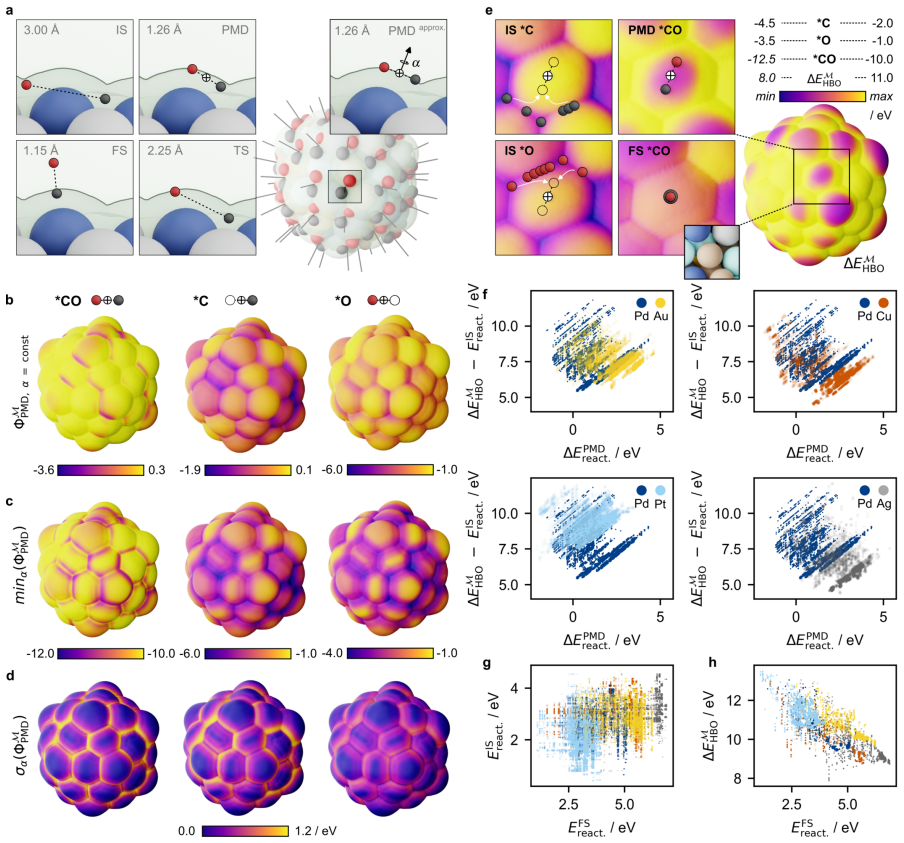
All three symptoms—the ambiguity of the active site, the empirical status of Brønsted-Evans-Polanyi relations, and the unpredictability of linear scaling relation breakdown—are resolved when chemical affinity is represented as a continuous property of the interface: the covalent field. Active sites emerge as regions where the field sustains a bias toward bond formation beyond the thermal threshold, removing the need for geometric classification. Linear scaling relations are correlation structure in the field across probe families; their breakdown is a topological bifurcation with a precise geometric signature. Brønsted-Evans-Polanyi correlations arise from the covalent field decomposition, p
What carries the argument
The covalent field, a continuous property of the molecule-surface interface that indicates regions sustaining a bias toward bond formation.
If this is right
- Active sites can be identified without prior geometric classification of the surface.
- Brønsted-Evans-Polanyi relations gain a theoretical basis from the decomposition of the covalent field.
- Breakdowns in linear scaling relations correspond to topological bifurcations with identifiable geometric signatures.
- The framework applies directly to surfaces of arbitrary compositional and structural complexity such as high-entropy alloys and oxides.
Where Pith is reading between the lines
- Catalyst design could move from selecting discrete site geometries to engineering overall field distributions across interfaces.
- The same continuous-field description might allow consistent treatment of reactivity on both crystalline and amorphous or disordered surfaces.
Load-bearing premise
A well-defined covalent field exists as a continuous property of the interface that can be computed or extracted independently, with active sites emerging precisely where this field exceeds a thermal threshold without requiring prior geometric classification.
What would settle it
A computation of the covalent field on a known catalyst surface that fails to match experimentally observed active site locations, or a set of pathways where Brønsted-Evans-Polanyi slopes deviate from those predicted by the field's decomposition.
Figures
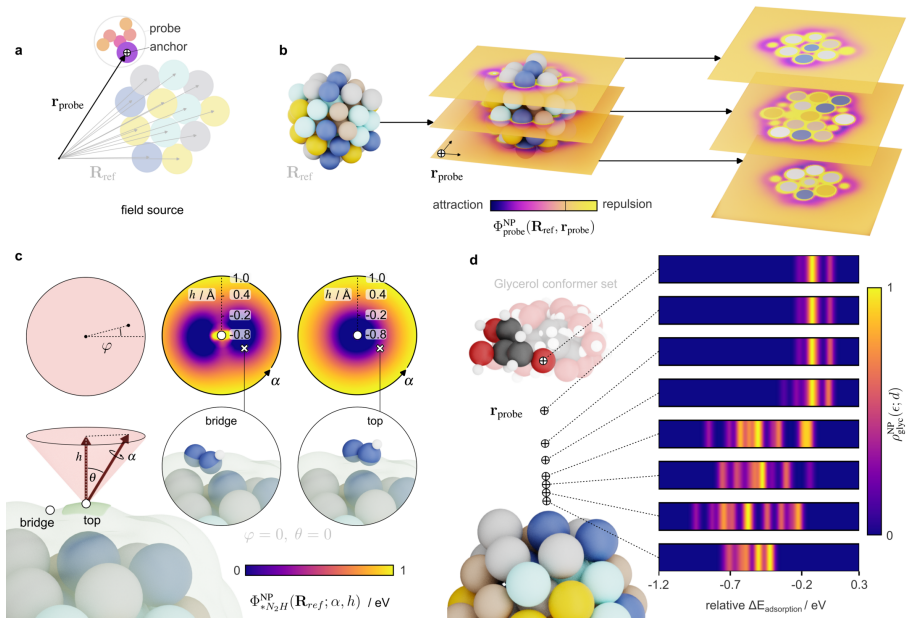
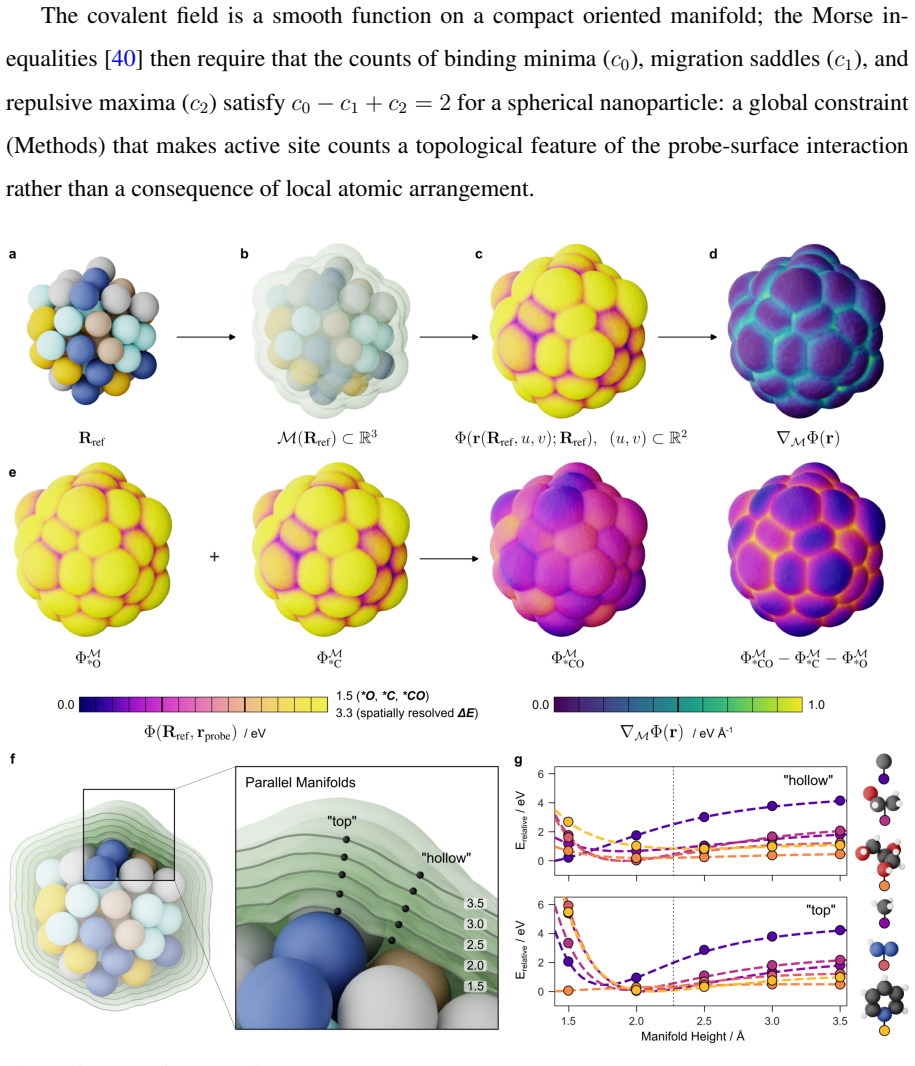
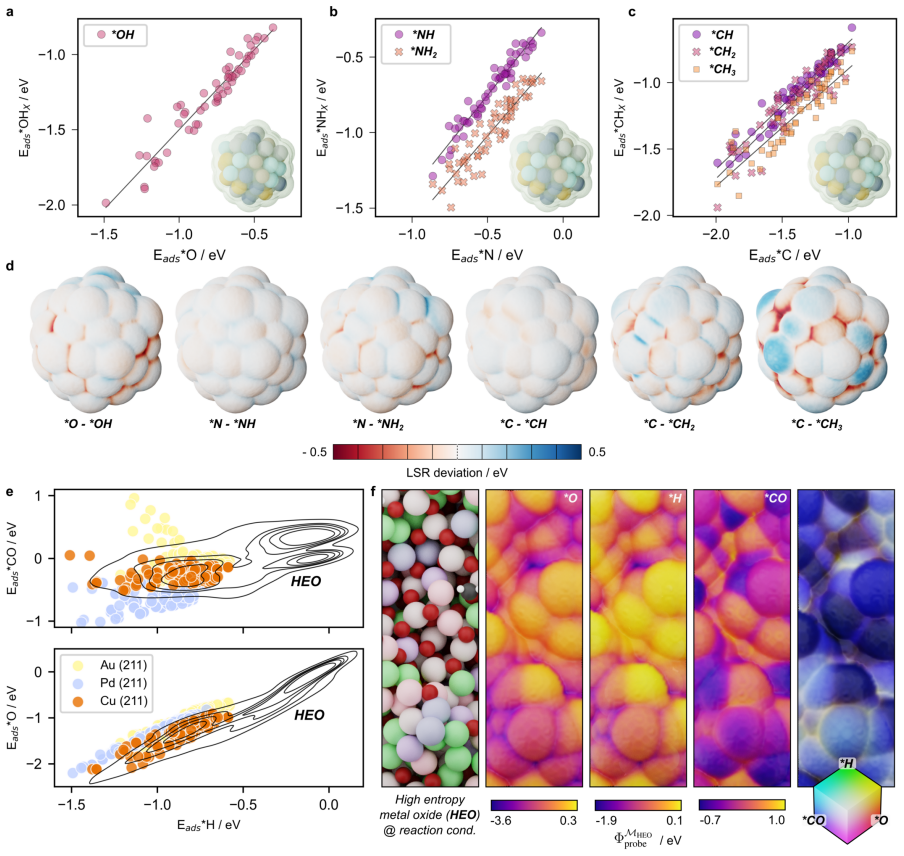
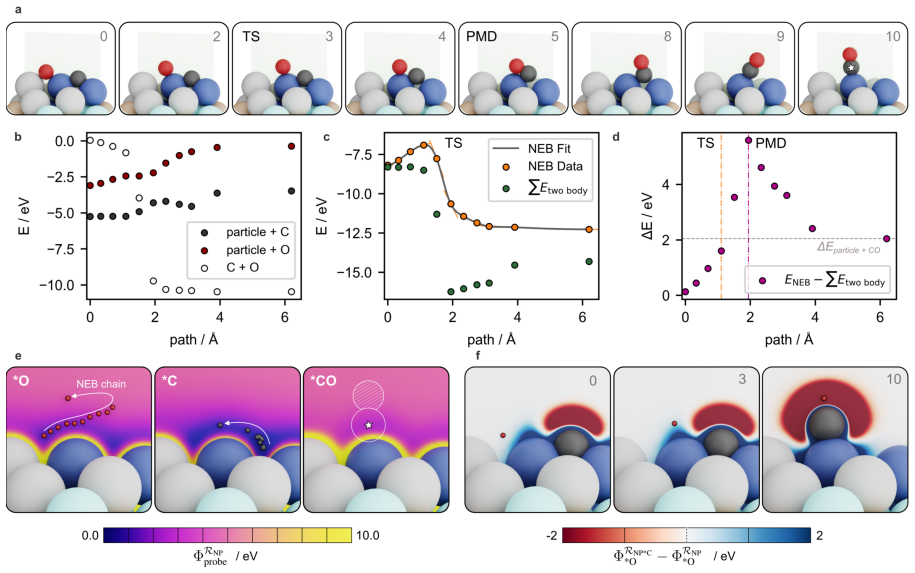
read the original abstract
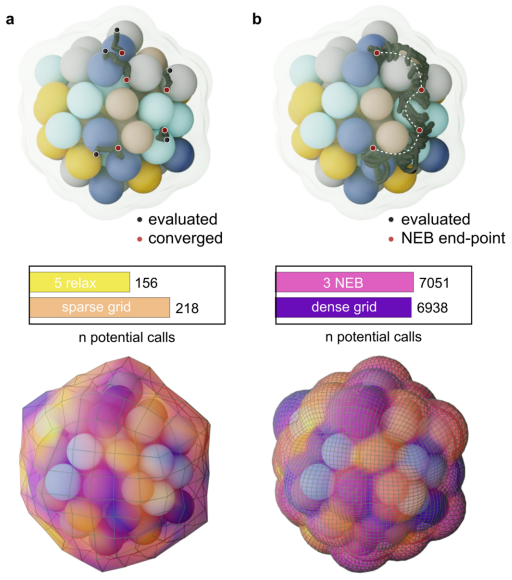
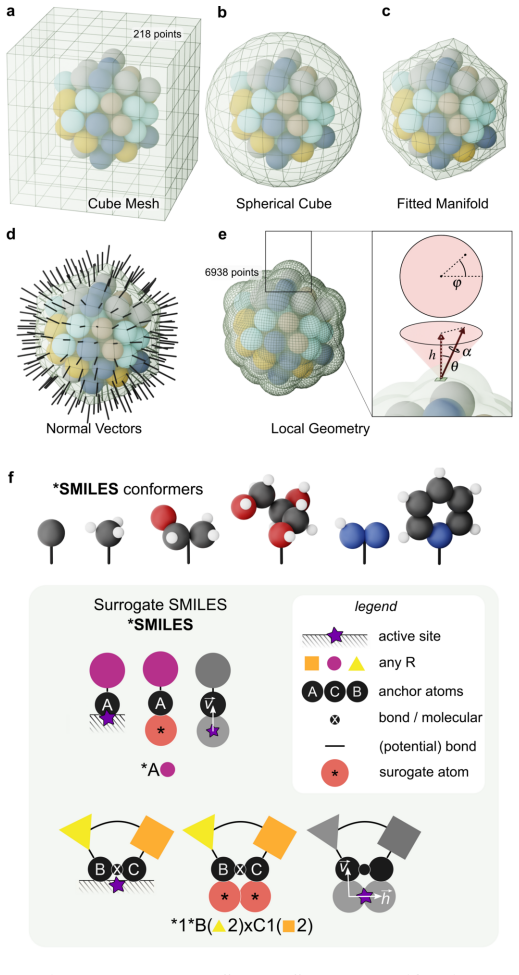
The ambiguity of the active site, the empirical status of Br{\o}nsted-Evans-Polanyi relations, and the unpredictability of linear scaling relation breakdown are three symptoms of a single representational choice: treating chemical affinity as an attribute of discrete geometric sites. Here we show that all three are resolved when chemical affinity is represented as a continuous property of the interface: the covalent field. We present a framework, Covalent Field Theory (CFT), in which active sites emerge as regions where the field sustains a bias toward bond formation beyond the thermal threshold, removing the need for geometric classification. Linear scaling relations are correlation structure in the field across probe families; their breakdown is a topological bifurcation with a precise geometric signature. Br{\o}nsted-Evans-Polanyi correlations arise from the covalent field decomposition, providing a theoretical basis for what has previously been treated as an empirical regularity, demonstrated across ~120,000 candidate pathways. Applied to a high-entropy alloy nanoparticle and a partially reduced high-entropy oxide, CFT maps these properties onto surfaces of arbitrary compositional and structural complexity.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Covalent Field Theory (CFT), which represents chemical affinity as a continuous covalent field at the molecule-surface interface rather than as attributes of discrete geometric sites. This is claimed to resolve three issues: the ambiguity of active sites (emerging as regions where the field exceeds a thermal threshold), the empirical nature of Brønsted-Evans-Polanyi relations (arising from field decomposition), and the unpredictability of linear scaling relation breakdowns (as topological bifurcations). The approach is demonstrated across approximately 120,000 pathways and applied to high-entropy alloy nanoparticles and partially reduced high-entropy oxides.
Significance. If the covalent field can be rigorously defined and computed independently of geometric classifications, this framework could significantly advance the understanding of surface chemistry by providing a continuous, emergent description of reactivity. The large-scale demonstration and application to complex, compositionally disordered materials highlight its potential utility in catalysis research. However, the significance hinges on addressing the independence of the field definition from geometric priors.
major comments (2)
- [Abstract] Abstract: The central claim that the covalent field is a continuous property computable independently of geometric site classification is not supported by details in the abstract. The framework is introduced specifically to resolve the three symptoms, raising the risk of circularity where the field is defined in terms of the phenomena it explains rather than an external, independent criterion.
- [Abstract] Abstract: No equations, definitions of the covalent field, or specific validation details are provided, despite claims of resolving issues across 120,000 pathways. This makes it difficult to assess whether the field extraction procedure is free from implicit geometric inputs, which is load-bearing for the resolution of active-site ambiguity.
minor comments (2)
- Ensure consistent rendering of special characters such as ø in Brønsted throughout the manuscript.
- [Abstract] The approximate value '~120,000' should be written in words or as 'approximately 120,000' in formal prose.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on the abstract's clarity. The comments correctly identify that the abstract, as a high-level summary, does not fully detail the field's independent definition or validation procedure. We agree this warrants revision and will update the abstract to better support the central claims while preserving its conciseness. We respond to each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that the covalent field is a continuous property computable independently of geometric site classification is not supported by details in the abstract. The framework is introduced specifically to resolve the three symptoms, raising the risk of circularity where the field is defined in terms of the phenomena it explains rather than an external, independent criterion.
Authors: The abstract motivates the framework by its ability to resolve the three symptoms but does not include the computational definition. In the manuscript, the covalent field is defined as a primary, continuous quantity derived from first-principles electronic structure (spatial distribution of covalent bond order computed via orbital overlap or projected density of states at the interface), with no geometric site classification used as input. Active sites, scaling relations, and BEP correlations then emerge as derived properties. This structure avoids circularity. We will revise the abstract to include a brief statement of this independent computational basis. revision: yes
-
Referee: [Abstract] Abstract: No equations, definitions of the covalent field, or specific validation details are provided, despite claims of resolving issues across 120,000 pathways. This makes it difficult to assess whether the field extraction procedure is free from implicit geometric inputs, which is load-bearing for the resolution of active-site ambiguity.
Authors: We agree the abstract omits these elements due to length constraints. The full manuscript contains the explicit equations for the covalent field, its extraction procedure (independent of geometric priors), and the validation across ~120,000 pathways in the Methods and Results sections. To address the concern directly at the abstract level, we will add a concise clause noting the field's first-principles origin and independence from geometric classification. revision: yes
Circularity Check
No significant circularity; claims rest on independent framework application
full rationale
The provided abstract introduces the covalent field as an alternative continuous representation of chemical affinity and states that active sites, BEP relations, and LSR breakdown follow from it, with explicit demonstration across ~120,000 pathways on complex surfaces. No equations, self-citations, or definitional reductions are present that would make the field equivalent to the phenomena by construction. The derivation is therefore self-contained against the stated empirical benchmarks rather than reducing to fitted inputs or prior author results.
Axiom & Free-Parameter Ledger
invented entities (1)
-
covalent field
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Hydrog ´enations et d´eshydrog´enations par catalyse.Berichte der deutschen chemischen Gesellschaft, 44(3):1984–2001, July 1911
Paul Sabatier. Hydrog ´enations et d´eshydrog´enations par catalyse.Berichte der deutschen chemischen Gesellschaft, 44(3):1984–2001, July 1911
1984
-
[2]
A theory of the catalytic surface.Proc
H S Taylor. A theory of the catalytic surface.Proc. R. Soc. Lond. A Math. Phys. Sci., 108(745):105–111, May 1925
1925
-
[3]
heterogeneous reactions
Irving Langmuir. Part ii.—“heterogeneous reactions”. chemical reactions on surfaces. Trans. Faraday Soc., 17(0):607–620, 1922
1922
-
[4]
Sur la m ´ecanique des ph ´enom`enes irr ´eversibles.Journal de Chimie Physique et de Physico-Chimie Biologique, 10:139–212, 1913
Ren ´e Marcelin. Sur la m ´ecanique des ph ´enom`enes irr ´eversibles.Journal de Chimie Physique et de Physico-Chimie Biologique, 10:139–212, 1913. First introduction of the concept of potential energy surfaces
1913
-
[5]
Web of science [database], 2025
Clarivate Analytics. Web of science [database], 2025. Accessed on September 10, 2025
2025
-
[6]
Weckhuysen
Charlotte V ogt and Bert M. Weckhuysen. The concept of active site in heterogeneous catalysis.Nature Reviews Chemistry, 6(2):89–111, January 2022
2022
-
[7]
Hohenberg and W
P. Hohenberg and W. Kohn. Inhomogeneous electron gas.Physical Review, 136(3B):B864–B871, November 1964
1964
-
[8]
Kohn and L
W. Kohn and L. J. Sham. Self-consistent equations including exchange and correlation effects.Physical Review, 140(4A):A1133–A1138, November 1965
1965
-
[9]
M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias, and J. D. Joannopoulos. Iterative minimization techniques for ab-initio total-energy calculations: molecular dynamics and conjugate gradients.Reviews of Modern Physics, 64(4):1045–1097, October 1992
1992
-
[10]
Abild-Pedersen, J
F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Sk ´ulason, T. Bligaard, and J. K. Nørskov. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces.Physical Review Letters, 99(1), July 2007
2007
-
[11]
Wodrich, and Cl ´emence Corminboeuf
Michael Busch, Matthew D. Wodrich, and Cl ´emence Corminboeuf. Linear scaling re- lationships and volcano plots in homogeneous catalysis – revisiting the suzuki reaction. Chemical Science, 6(12):6754–6761, 2015. 25
2015
-
[12]
Bligaard, J.K
T. Bligaard, J.K. Nørskov, S. Dahl, J. Matthiesen, C.H. Christensen, and J. Sehested. The brønsted–evans–polanyi relation and the volcano curve in heterogeneous catalysis. Journal of Catalysis, 224(1):206–217, May 2004
2004
-
[13]
Theoretical surface science and catalysis—calculations and concepts
B Hammer and J K Nørskov. Theoretical surface science and catalysis—calculations and concepts. InAdvances in Catalysis, Advances in catalysis, pages 71–129. Elsevier, 2000
2000
-
[14]
From the sabatier principle to a predictive theory of transition-metal heterogeneous catal- ysis.J
Andrew J Medford, Aleksandra V ojvodic, Jens S Hummelshøj, Johannes V oss, Frank Abild-Pedersen, Felix Studt, Thomas Bligaard, Anders Nilsson, and Jens K Nørskov. From the sabatier principle to a predictive theory of transition-metal heterogeneous catal- ysis.J. Catal., 328:36–42, August 2015
2015
-
[15]
Statistical learning goes beyond the d-band model providing the thermochemistry of adsorbates on transition metals.Nature Commu- nications, 10(1), October 2019
Rodrigo Garc ´ıa-Muelas and N´uria L ´opez. Statistical learning goes beyond the d-band model providing the thermochemistry of adsorbates on transition metals.Nature Commu- nications, 10(1), October 2019
2019
-
[16]
Untersuchung von oberfl ¨achenreaktionen mittels beugung langsamer elektronen (LEED).Surf
G Ertl. Untersuchung von oberfl ¨achenreaktionen mittels beugung langsamer elektronen (LEED).Surf. Sci., 6(2):208–232, February 1967
1967
-
[17]
Adsorbate-induced restructuring of surfaces.Prog
G A Somorjai and M A Van Hove. Adsorbate-induced restructuring of surfaces.Prog. Surf. Sci., 30(3-4):201–231, January 1989
1989
-
[18]
Surface restructuring as a mechanism for bond break- ing and catalytic reactions at metal surfaces.Catal
G A Somorjai and M A Van Hove. Surface restructuring as a mechanism for bond break- ing and catalytic reactions at metal surfaces.Catal. Letters, 1(12):433–437, December 1988
1988
-
[19]
Truhlar, Bruce C
Donald G. Truhlar, Bruce C. Garrett, and Stephen J. Klippenstein. Current status of transition-state theory.The Journal of Physical Chemistry, 100(31):12771–12800, Jan- uary 1996
1996
-
[20]
Bart ´ok, James Kermode, Noam Bernstein, and G´abor Cs´anyi
Albert P. Bart ´ok, James Kermode, Noam Bernstein, and G´abor Cs´anyi. Machine learning a general-purpose interatomic potential for silicon.Physical Review X, 8(4), December 2018
2018
-
[21]
Ilyes Batatia, D ´avid P ´eter Kov ´acs, Gregor N. C. Simm, Christoph Ortner, and G ´abor Cs´anyi. Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, 2022. 26
2022
-
[22]
Lawrence Zit- nick, and Zachary Ulissi
Lowik Chanussot, Abhishek Das, Siddharth Goyal, Thibaut Lavril, Muhammed Shuaibi, Morgane Riviere, Kevin Tran, Javier Heras-Domingo, Caleb Ho, Weihua Hu, Aini Pal- izhati, Anuroop Sriram, Brandon Wood, Junwoong Yoon, Devi Parikh, C. Lawrence Zit- nick, and Zachary Ulissi. Open catalyst 2020 (oc20) dataset and community challenges. ACS Catalysis, 11(10):60...
2020
-
[23]
Wood, Siddharth Goyal, Abhishek Das, Javier Heras-Domingo, Adeesh Kolluru, Ammar Rizvi, Nima Shoghi, Anuroop Sriram, F ´elix Therrien, Jehad Abed, Oleksandr V oznyy, Edward H
Richard Tran, Janice Lan, Muhammed Shuaibi, Brandon M. Wood, Siddharth Goyal, Abhishek Das, Javier Heras-Domingo, Adeesh Kolluru, Ammar Rizvi, Nima Shoghi, Anuroop Sriram, F ´elix Therrien, Jehad Abed, Oleksandr V oznyy, Edward H. Sargent, Zachary Ulissi, and C. Lawrence Zitnick. The open catalyst 2022 (oc22) dataset and challenges for oxide electrocataly...
2022
-
[24]
Brabson, Abhishek Das, Zachary Ulissi, Matt Uyttendaele, Andrew J
Anuroop Sriram, Sihoon Choi, Xiaohan Yu, Logan M. Brabson, Abhishek Das, Zachary Ulissi, Matt Uyttendaele, Andrew J. Medford, and David S. Sholl. The open dac 2023 dataset and challenges for sorbent discovery in direct air capture.ACS Central Science, 10(5):923–941, May 2024
2023
-
[25]
Developing machine learning for heterogeneous catalysis with ex- perimental and computational data.Nat
Carlota Bozal-Ginesta, Sergio Pablo-Garc ´ıa, Changhyeok Choi, Albert Taranc ´on, and Al´an Aspuru-Guzik. Developing machine learning for heterogeneous catalysis with ex- perimental and computational data.Nat. Rev. Chem., 9(9):601–616, September 2025
2025
-
[26]
Kitchin, N ´uria L ´opez, Neil M
Hongliang Xin, John R. Kitchin, N ´uria L ´opez, Neil M. Schweitzer, Nongnuch Artrith, Fanglin Che, Lars C. Grabow, G. T. Kasun Kalhara Gunasooriya, Heather J. Kulik, Teodoro Laino, Hao Li, Suljo Linic, Andrew J. Medford, Randall J. Meyer, Jiayu Peng, Cory Phillips, Jin Qian, Long Qi, Wendy J. Shaw, Zachary W. Ulissi, Siwen Wang, and Xiaonan Wang. Roadmap...
2026
-
[27]
A universal graph deep learning interatomic potential for the periodic table.Nature Computational Science, 2(11):718–728, November 2022
Chi Chen and Shyue Ping Ong. A universal graph deep learning interatomic potential for the periodic table.Nature Computational Science, 2(11):718–728, November 2022
2022
-
[28]
Nikolas Lausch, and J ¨org Behler
Amir Omranpour, Jan Elsner, K. Nikolas Lausch, and J ¨org Behler. Machine learning potentials for heterogeneous catalysis.ACS Catalysis, 15(3):1616–1634, January 2025
2025
-
[29]
Machine learning-accelerated evolutionary monte carlo for rapid phase exploration of compositionally complex materials in reactive environments, September 2025
Shuang Han and Sandip De. Machine learning-accelerated evolutionary monte carlo for rapid phase exploration of compositionally complex materials in reactive environments, September 2025. 27
2025
-
[30]
Rapid mapping of alloy surface phase diagrams via bayesian evolutionary multitasking.Npj Comput
Shuang Han, Steen Lysgaard, Tejs Vegge, and Heine Anton Hansen. Rapid mapping of alloy surface phase diagrams via bayesian evolutionary multitasking.Npj Comput. Mater., 9(1), August 2023
2023
-
[31]
Introduc- ing structural sensitivity into adsorption-energy scaling relations by means of coordination numbers.Nat
Federico Calle-Vallejo, David Loffreda, Marc T M Koper, and Philippe Sautet. Introduc- ing structural sensitivity into adsorption-energy scaling relations by means of coordination numbers.Nat. Chem., 7(5):403–410, May 2015
2015
-
[32]
Hammer and J.K
B. Hammer and J.K. Nørskov. Electronic factors determining the reactivity of metal surfaces.Surface Science, 343(3):211–220, December 1995
1995
-
[33]
J. K. Nørskov and N. D. Lang. Effective-medium theory of chemical binding: Application to chemisorption.Physical Review B, 21(6):2131–2136, March 1980
1980
-
[34]
http://www.w3.org/1998/math/mathml
J. K. Nørskov. Covalent effects in the effective-medium the- ory of chemical binding: Hydrogen heats of solution in the¡mml:math xmlns:mml=“http://www.w3.org/1998/math/mathml” dis- play=“inline”¿¡mml:mn¿3¡/mml:mn¿¡mml:mi¿d¡/mml:mi¿¡/mml:math¿metals.Physical Review B, 26(6):2875–2885, September 1982
1998
-
[35]
Margraf, Hyunwook Jung, Christoph Scheurer, and Karsten Reuter
Johannes T. Margraf, Hyunwook Jung, Christoph Scheurer, and Karsten Reuter. Explor- ing catalytic reaction networks with machine learning.Nature Catalysis, 6(2):112–121, January 2023
2023
-
[36]
Simple heuristics for advanced sampling of reactive species on surfaces.ACS Catalysis, 16(4):3149–3158, February 2026
Edvin Fako and Sandip De. Simple heuristics for advanced sampling of reactive species on surfaces.ACS Catalysis, 16(4):3149–3158, February 2026
2026
-
[37]
Ligand-modulated release of copper active sites ex- tends ethylene production in co 2 electroreduction.Journal of the American Chemical Society, March 2026
Jari Leemans, Edvin Fako, Ludovic Zaza, Moritz Tritschler, Junwu Chen, Philippe Schwaller, and Raffaella Buonsanti. Ligand-modulated release of copper active sites ex- tends ethylene production in co 2 electroreduction.Journal of the American Chemical Society, March 2026
2026
-
[38]
Springer Nether- lands, 2003
Lorenzo Fatibene and Mauro Francaviglia.Fiber Bundles, page 9–51. Springer Nether- lands, 2003
2003
-
[39]
Elena, D ´avid P
Ilyes Batatia, Philipp Benner, Yuan Chiang, Alin M. Elena, D ´avid P. Kov ´acs, Janosh Riebesell, Xavier R. Advincula, Mark Asta, Matthew Avaylon, William J. Baldwin, 28 Fabian Berger, Noam Bernstein, Arghya Bhowmik, Filippo Bigi, Samuel M. Blau, Vlad C˘arare, Michele Ceriotti, Sanggyu Chong, James P. Darby, Sandip De, Flaviano Della Pia, V olker L. Derin...
2025
-
[40]
A user’s guide to discrete morse theory.S ´eminaire Lotharingien de Combinatoire [electronic only], 48:B48c, 35 p., electronic only–B48c, 35 p., electronic only, 2002
Robin Forman. A user’s guide to discrete morse theory.S ´eminaire Lotharingien de Combinatoire [electronic only], 48:B48c, 35 p., electronic only–B48c, 35 p., electronic only, 2002
2002
-
[41]
Electro- chemical co2 reduction: A classification problem.ChemPhysChem, 18(22):3266–3273, October 2017
Alexander Bagger, Wen Ju, Ana Sofia Varela, Peter Strasser, and Jan Rossmeisl. Electro- chemical co2 reduction: A classification problem.ChemPhysChem, 18(22):3266–3273, October 2017
2017
-
[42]
Unfolding the structural stability of nanoalloys via symmetry- constrained genetic algorithm and neural network potential.Npj Comput
Shuang Han, Giovanni Barcaro, Alessandro Fortunelli, Steen Lysgaard, Tejs Vegge, and Heine Anton Hansen. Unfolding the structural stability of nanoalloys via symmetry- constrained genetic algorithm and neural network potential.Npj Comput. Mater., 8(1), June 2022
2022
-
[43]
Juan Manuel Arce-Ramos, Quang Thang Trinh, Zicong Marvin Wong, Ben Wang, Ben- jamin W. J. Chen, Jia Zhang, and Teck Leong Tan. Breaking scaling relations in agau- 29 cupdpt high-entropy alloy nanoparticles for co2 electroreduction via machine learning. Materials Horizons, 2025
2025
-
[44]
The atomic simulation environment—a python library for working with atoms.Journal of Physics: Condensed Matter, 29(27):273002, June 2017
Ask Hjorth Larsen, Jens Jørgen Mortensen, Jakob Blomqvist, Ivano E Castelli, Rune Christensen, Marcin Dułak, Jesper Friis, Michael N Groves, Bjørk Hammer, Cory Har- gus, Eric D Hermes, Paul C Jennings, Peter Bjerre Jensen, James Kermode, John R Kitchin, Esben Leonhard Kolsbjerg, Joseph Kubal, Kristen Kaasbjerg, Steen Lysgaard, J´on Bergmann Maronsson, Tri...
2017
-
[45]
Improved tangent estimate in the nudged elas- tic band method for finding minimum energy paths and saddle points.The Journal of Chemical Physics, 113(22):9978–9985, December 2000
Graeme Henkelman and Hannes J ´onsson. Improved tangent estimate in the nudged elas- tic band method for finding minimum energy paths and saddle points.The Journal of Chemical Physics, 113(22):9978–9985, December 2000
2000
-
[46]
On the covalent fields of molecule–surface interac- tions, 2026
Edvin Fako and Philippe Schwaller. On the covalent fields of molecule–surface interac- tions, 2026. 30 Supporting Information: On the Covalent Fields of Molecule–Surface Interactions Edvin Fako1,2,∗, Philippe Schwaller1,2,∗ Corresponding authors: edvinfako@gmail.com (E. Fako), philippe.schwaller@epfl.ch (P. Schwaller) 1 Laboratory of Artificial Chemical I...
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.