Contextualizing Biological Language Models across Modalities via Logit-Space Contrastive Alignment
Pith reviewed 2026-06-26 21:29 UTC · model grok-4.3
The pith
LOGICA aligns biological language models in logit space to add context from ligands or drugs while preserving the original per-token likelihood interface.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
LOGICA performs contrastive learning directly in output-logit space with gated cross-modal adapters that interface with each model's native token head, producing context-conditioned token probabilities that improve mutation-local variant ranking on protein-ligand, TCR-peptide, and drug-resistance tasks while preserving the pretrained per-token likelihood interface and requiring no shared tokenizer.
What carries the argument
Gated cross-modal adapters that map contextual inputs into adjustments of each base model's logit outputs, enabling contrastive alignment of token probabilities across modalities.
If this is right
- Mutation-local variant ranking reduces to direct comparison of context-conditioned token likelihoods at the perturbed site.
- Models with distinct vocabularies can be aligned for joint prediction using only sparse paired examples.
- The native token-level interface remains available for both mechanistic interpretation and sequence generation.
- AUC on held-out-gene single-mutation drug-resistance prediction rises from near-random latent baselines of ~0.55 to ~0.65.
Where Pith is reading between the lines
- The same logit-space alignment could be tested on additional modalities such as gene-expression or metabolite contexts to check whether the gains generalize beyond the three tasks studied.
- Working in probability space rather than embedding space may reduce the data needed to combine separately pretrained biological models.
- The approach could be applied to score multi-mutation combinations under drug pressure without retraining the base language model.
Load-bearing premise
Gated adapters can align logits across models with different vocabularies using only sparse paired data without distorting the original per-token probability distributions.
What would settle it
On a new held-out set of single-mutation drug-resistance cases, if the context-conditioned token likelihoods produced by LOGICA rank true resistant variants no higher than the uncontextualized base model, the central claim would be falsified.
Figures

read the original abstract
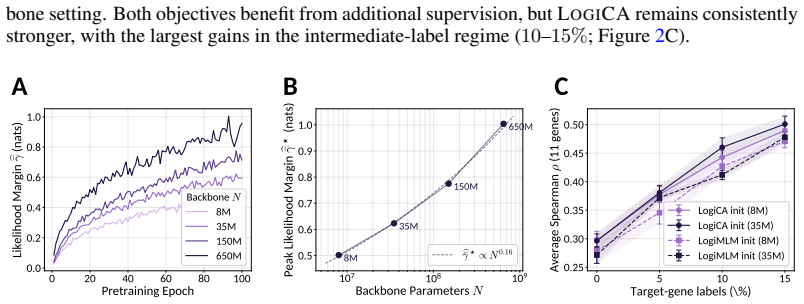
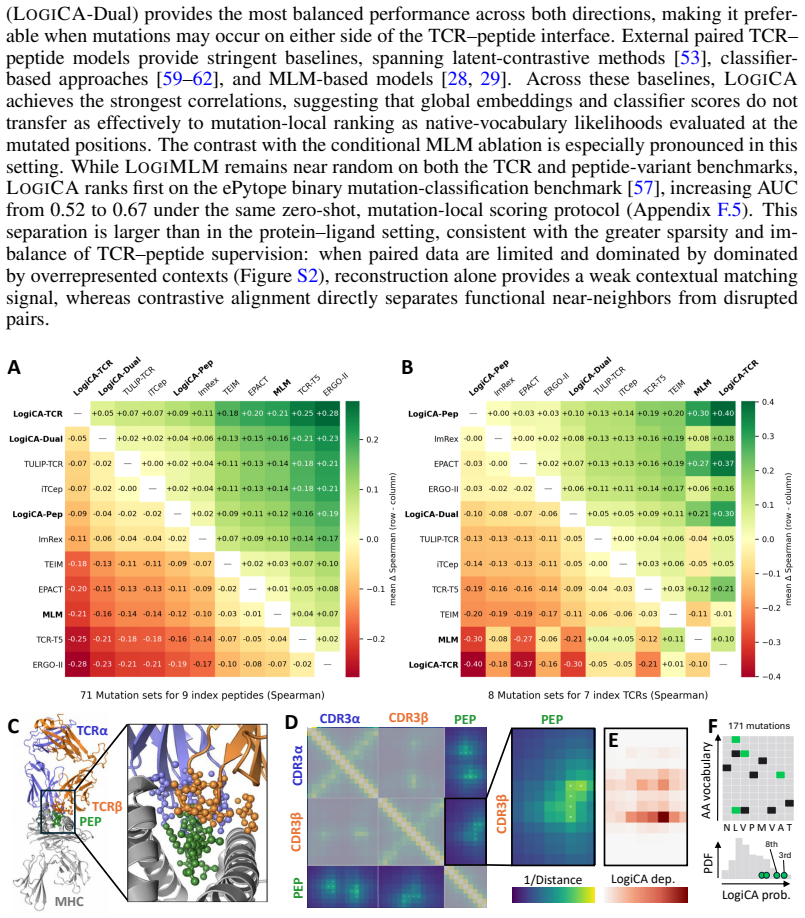
Pretrained biological language models expose per-token probability distributions through masked-token prediction, providing the likelihood interface central to sequence design, variant scoring, and mechanistic interpretation. Yet these distributions are learned from broad unlabeled corpora and are not naturally conditioned on task-specific biological contexts such as interaction partners, cellular environments, or therapeutic interventions. Existing contextual matching methods often distort this interface through pooled embeddings, contrastive latent spaces, or task-specific prediction heads. We introduce LOGICA (Logit-space Contrastive Alignment), a framework for context-conditioned prediction that performs contrastive learning directly in output-logit space. Using gated cross-modal adapters compatible with each model's native token head, LOGICA preserves the pretrained likelihood interface and converts contextualized token log-likelihoods into matching scores. Alignment is defined through context-sensitive token probabilities rather than proximity in a shared embedding space, enabling learning from sparse paired data across models with distinct vocabularies, without a shared tokenizer or decoder. LOGICA is particularly effective for mutation-local variant ranking, where comparisons reduce to context-conditioned likelihoods of mutant tokens at perturbed sites. Across protein--ligand binding, TCR--peptide activity, and drug-conditioned resistance prediction, LOGICA improves over prior state-of-the-art methods, including matched latent-contrastive and conditional MLM baselines, while retaining a token-level interface for interpretation and generation. On held-out-gene single-mutation drug-resistance prediction, LOGICA improves AUC from near-random latent-space baselines of $\sim$0.55 to $\sim$0.65.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces LOGICA, a logit-space contrastive alignment framework that uses gated cross-modal adapters to condition pretrained biological language models on task-specific contexts (e.g., ligands, peptides, drugs) while preserving the native per-token likelihood interface. It reports AUC gains over latent-space and conditional-MLM baselines on protein-ligand binding, TCR-peptide activity, and held-out-gene single-mutation drug-resistance prediction (0.55 to 0.65), with the alignment performed directly on context-sensitive token probabilities rather than embeddings and without requiring a shared tokenizer.
Significance. If the preservation of the original per-token likelihood surface is empirically validated, the method would provide a practical route to contextualized variant scoring and generation that retains interpretability advantages of the pretrained token heads. The held-out-gene evaluation and cross-vocabulary compatibility are positive design choices that strengthen the generalization claim.
major comments (2)
- [Abstract] Abstract: the central claim that gated adapters 'preserve the pretrained likelihood interface' and 'convert contextualized token log-likelihoods into matching scores' without distortion is load-bearing for the interpretation/generation benefit, yet no direct metric (KL divergence, rank correlation, or calibration error) is reported comparing pre- and post-adapter per-token log-probabilities on held-out unpaired sequences outside the paired training distribution.
- [Abstract] Abstract: the reported AUC improvement (∼0.55 to ∼0.65) on held-out-gene drug-resistance prediction lacks accompanying details on training-set sizes, number of paired examples, statistical significance, error bars, or ablation controls on the adapter architecture, making it impossible to assess whether the gain arises from logit-space alignment or from other factors.
minor comments (1)
- [Abstract] Abstract: the phrase 'near-random latent-space baselines of ∼0.55' should be replaced by the exact baseline values and the precise latent-space method used for comparison.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that gated adapters 'preserve the pretrained likelihood interface' and 'convert contextualized token log-likelihoods into matching scores' without distortion is load-bearing for the interpretation/generation benefit, yet no direct metric (KL divergence, rank correlation, or calibration error) is reported comparing pre- and post-adapter per-token log-probabilities on held-out unpaired sequences outside the paired training distribution.
Authors: We agree that direct quantitative validation of likelihood preservation on held-out unpaired sequences is a valuable addition. The gated adapter design inserts a residual connection from the original logits, which by construction leaves the token head unchanged, but we acknowledge the absence of explicit metrics such as KL divergence or rank correlation in the current version. In revision we will add these metrics (KL, Spearman correlation of token ranks, and expected calibration error) computed on held-out sequences drawn from the pretraining distribution, reported in a new results subsection and referenced from the abstract. revision: yes
-
Referee: [Abstract] Abstract: the reported AUC improvement (∼0.55 to ∼0.65) on held-out-gene drug-resistance prediction lacks accompanying details on training-set sizes, number of paired examples, statistical significance, error bars, or ablation controls on the adapter architecture, making it impossible to assess whether the gain arises from logit-space alignment or from other factors.
Authors: We will revise the abstract to incorporate the requested details: approximate number of paired examples for the drug-resistance task, error bars from repeated runs, and a statement of statistical significance. Ablation results on adapter components (gating, contrastive objective) are already present in the supplementary material; we will add an explicit cross-reference in the abstract. These changes will allow readers to evaluate the source of the observed improvement without altering the core claims. revision: yes
Circularity Check
No circularity; new training procedure with independent empirical results
full rationale
The paper introduces LOGICA as a novel contrastive alignment framework operating directly in output-logit space via gated adapters. No equations, derivations, or self-citations in the provided text reduce the reported AUC gains (0.55 to 0.65) or the preservation of the token-level interface to quantities defined by construction from the same fitted parameters or prior self-referential results. The method is presented as an independent training procedure whose value is assessed via downstream task performance on held-out data, without any load-bearing step that renames a fit as a prediction or imports uniqueness via author-overlapping citations. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Evolutionary-scale prediction of atomic-level protein structure with a language model,
Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, Allan Dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Salvatore Candido, and Alexander Rives. Evolutionary-scale prediction of atomic- level protein structure with a language model.Science, 379(6637):1123–1130, 2023. doi:...
-
[2]
Adam J Riesselman, John B Ingraham, and Debora S Marks. Deep generative models of genetic variation capture the effects of mutations.Nature methods, 15(10):816–822, 2018. doi: 10.1038/s41592-018-0138-4. URLhttps://doi.org/10.1038/s41592-018-0138-4
-
[3]
Joshua Meier, Roshan Rao, Robert Verkuil, Jason Liu, Tom Sercu, and Alexander Rives. Lan- guage models enable zero-shot prediction of the effects of mutations on protein function. In Advances in Neural Information Processing Systems, volume 34, pages 29287–29303, 2021. doi: 10.1101/2021.07.09.450648. URLhttps://doi.org/10.1101/2021.07.09.450648
-
[4]
Brian L. Hie, Varun R. Shanker, Duo Xu, Theodora U. J. Bruun, Payton A. Weiden- bacher, Shaogeng Tang, Wesley Wu, John E. Pak, and Peter S. Kim. Efficient evolution of human antibodies from general protein language models.Nature Biotechnology, 42(2): 275–283, 2024. doi: 10.1038/s41587-023-01763-2. URLhttps://doi.org/10.1038/ s41587-023-01763-2
-
[5]
Hie, Kevin K
Brian L. Hie, Kevin K. Yang, and Peter S. Kim. Evolutionary velocity with protein language models predicts evolutionary dynamics of diverse proteins.Cell Systems, 13(4):274–285,
-
[6]
URLhttps://doi.org/10.1016/j.cels.2022
doi: 10.1016/j.cels.2022.01.003. URLhttps://doi.org/10.1016/j.cels.2022. 01.003
-
[7]
Atakan Y ¨uksel, Erva Ulusoy, Atabey ¨Unl¨u, and Tunca Do ˘gan. SELFormer: Molecular rep- resentation learning via SELFIES language models.Machine Learning: Science and Tech- nology, 4(2):025035, 2023. doi: 10.1088/2632-2153/acdb30. URLhttps://doi.org/10. 1088/2632-2153/acdb30
-
[8]
Hugo Dalla-Torre, Liam Gonzalez, Javier Mendoza-Revilla, Nicolas Lopez Carranza, Adam Henryk Grzywaczewski, Francesco Oteri, Christian Dallago, Evan Trop, Bernardo P. de Almeida, Hassan Sirelkhatim, Guillaume Richard, Marcin Skwark, Karim Beguir, Marie Lopez, and Thomas Pierrot. Nucleotide transformer: building and evaluating robust foun- dation models fo...
-
[9]
Claire Donnat and Elena Tuzhilina
Haotian Cui, Alejandro Tejada-Lapuerta, Maria Brbi ´c, Julio Saez-Rodriguez, Simona Cristea, Hani Goodarzi, Mohammad Lotfollahi, Fabian J Theis, and Bo Wang. Towards multimodal foundation models in molecular cell biology.Nature, 640(8059):623–633, 2025. doi: 10.1038/ s41586-025-08710-y. URLhttps://doi.org/10.1038/s41586-025-08710-y
-
[10]
Garyk Brixi, Matthew G. Durrant, Jerome Ku, Mohsen Naghipourfar, Michael Poli, Gwang- gyu Sun, Greg Brockman, Daniel Chang, Alison Fanton, Gabriel A. Gonzalez, Samuel H. King, David B. Li, Aditi T. Merchant, Eric Nguyen, Chiara Ricci-Tam, David W. Romero, Jonathan C. Schmok, Ali Taghibakhshi, Anton V orontsov, Brandon Yang, Myra Deng, Liv Gorton, Nam Nguy...
-
[11]
Pedro Tomaz da Silva, Alexander Karollus, Johannes Hingerl, Gihanna Galindez, Nils Wag- ner, Xavier Hernandez-Alias, Danny Incarnato, and Julien Gagneur. Nucleotide dependency analysis of genomic language models detects functional elements.Nature Genetics, 57: 2589–2602, 2025. doi: 10.1038/s41588-025-02347-3. URLhttps://doi.org/10.1038/ s41588-025-02347-3
-
[12]
Klivans, James Madigan Loy, Tianlong Chen, Qiang Liu, and Daniel Jesus Diaz
Chengyue Gong, Adam R. Klivans, James Madigan Loy, Tianlong Chen, Qiang Liu, and Daniel Jesus Diaz. Evolution-inspired loss functions for protein representation learning. In Proceedings of the 41st International Conference on Machine Learning (ICML), volume 235 ofProceedings of Machine Learning Research, pages 15893–15906. PMLR, 2024. URL https://proceedi...
2024
-
[13]
Wayment-Steele, Garyk Brixi, Hong Wang, David Kern, and Sergey Ovchinnikov
Zhidian Zhang, Hannah K. Wayment-Steele, Garyk Brixi, Hong Wang, David Kern, and Sergey Ovchinnikov. Protein language models learn evolutionary statistics of interacting se- quence motifs.Proceedings of the National Academy of Sciences, 121(45):e2406285121, 2024. doi: 10.1073/pnas.2406285121. URLhttps://doi.org/10.1073/pnas.2406285121
-
[14]
Greene, Subu Subramanian, Benjamin P
Ali Madani, Ben Krause, Eric R. Greene, Subu Subramanian, Benjamin P. Mohr, James M. Holton, Jose Luis Olmos Jr, Caiming Xiong, Zachary Z. Sun, Richard Socher, James S. Fraser, and Nikhil Naik. Large language models generate functional protein sequences across diverse families.Nature biotechnology, 41(8):1099–1106, 2023. doi: 10.1038/s41587-022-01618-2. U...
-
[15]
Sean R Johnson, Sarah Monaco, Kenneth Massie, and Zaid Syed. Generating novel protein sequences using gibbs sampling of masked language models.bioRxiv, pages 2021–01, 2021. doi: 10.1101/2021.01.26.428322. URLhttps://doi.org/10.1101/2021.01.26.428322
-
[16]
How to make the most of your masked language model for protein engineering
Calvin McCarter, Nick Bhattacharya, Sebastian W Ober, and Hunter Elliott. How to make the most of your masked language model for protein engineering.arXiv preprint arXiv:2603.10302, 2026. doi: 10.48550/arXiv.2603.10302. URLhttps://arxiv.org/abs/ 2603.10302
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2603.10302 2026
-
[17]
Josh Abramson, Jonas Adler, Jack Dunger, Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger, Lindsay Willmore, Andrew J. Ballard, Joshua Bambrick, Sebastian W. Boden- stein, David A. Evans, Chia-Chun Hung, Michael O’Neill, David Reiman, Kathryn Tunyasu- vunakool, Zachary Wu, Akvil ˙e ˇZemgulyt˙e, Eirini Arvaniti, Charles Beattie, Ottavia Bertol...
-
[18]
Yicheng Gao, Yuli Gao, Yuxiao Fan, Chengyu Zhu, Zhiting Wei, Chi Zhou, Guohui Chuai, Qinchang Chen, He Zhang, and Qi Liu. Pan-peptide meta learning for T-cell receptor–antigen binding recognition.Nature Machine Intelligence, 2023. doi: 10.1038/s42256-023-00619-3. URLhttps://doi.org/10.1038/s42256-023-00619-3
-
[19]
Boltz-2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, David Kwabi-Addo, Dominique Beaini, Tommi Jaakkola, and Regina Barzilay. Boltz-2: Towards accurate and efficient binding affinity prediction, 2025. URLhttps://doi.org/10.1101/2025.06.14.659707. bioRxiv preprint
-
[20]
Kexin Huang, Cao Xiao, Lucas M. Glass, and Jimeng Sun. MolTrans: Molecular in- teraction transformer for drug–target interaction prediction.Bioinformatics, 37(6):830– 836, 2021. doi: 10.1093/bioinformatics/btaa880. URLhttps://doi.org/10.1093/ bioinformatics/btaa880. 11
-
[21]
Deep contrastive learning enables genome-wide virtual screening.Science, 391(6781):eads9530, 2026
Yinjun Jia, Bowen Gao, Jiaxin Tan, Jiqing Zheng, Xin Hong, Wenyu Zhu, Haichuan Tan, Yuan Xiao, Liping Tan, Hongyi Cai, Yanwen Huang, Zhiheng Deng, Xiangwei Wu, Yue Jin, Yafei Yuan, Jiekang Tian, Wei He, Weiying Ma, Yaqin Zhang, Lei Liu, Chuangye Yan, Wei Zhang, and Yanyan Lan. Deep contrastive learning enables genome-wide virtual screening.Science, 391(67...
-
[22]
Rohit Singh, Samuel Sledzieski, Bryan Bryson, Lenore Cowen, and Bonnie Berger. Con- trastive learning in protein language space predicts interactions between drugs and protein targets.Proceedings of the National Academy of Sciences, 120(24):e2220778120, 2023. doi: 10.1073/pnas.2220778120. URLhttps://doi.org/10.1073/pnas.2220778120
-
[23]
Peizhen Bai, Filip Miljkovi ´c, Bino John, and Haiping Lu. Interpretable bilinear attention net- work with domain adaptation improves drug–target prediction.Nature Machine Intelligence, 5 (2):126–136, 2023. doi: 10.1038/s42256-022-00605-1. URLhttps://doi.org/10.1038/ s42256-022-00605-1
-
[24]
Sizhe Liu, Yuchen Liu, Haofeng Xu, Jun Xia, and Stan Z. Li. SP-DTI: subpocket-informed transformer for drug–target interaction prediction.Bioinformatics, 41(3):btaf011, 03 2025. ISSN 1367-4811. doi: 10.1093/bioinformatics/btaf011. URLhttps://doi.org/10.1093/ bioinformatics/btaf011
-
[25]
Qinze Yu, Chang Zhou, Jiyue Jiang, Xiangyu Shi, and Yu Li. GS-DTI: A graph-structure-aware framework leveraging large language models for drug–target interaction prediction.Bioinfor- matics, 41(8):btaf445, 08 2025. ISSN 1367-4811. doi: 10.1093/bioinformatics/btaf445. URL https://doi.org/10.1093/bioinformatics/btaf445
-
[26]
Weber, Ella Barkan, Simona Rabinovici-Cohen, Sagi Polaczek, Ido Amos, et al
Yoel Shoshan, Moshiko Raboh, Michal Ozery-Flato, Vadim Ratner, Alex Golts, Jeffrey K. Weber, Ella Barkan, Simona Rabinovici-Cohen, Sagi Polaczek, Ido Amos, et al. MAMMAL – molecular aligned multi-modal architecture and language for biomedical discovery.npj Drug Discovery, 2026. doi: 10.1038/s44386-026-00047-4. URLhttps://doi.org/10.1038/ s44386-026-00047-4
-
[27]
Varun Ullanat, Bowen Jing, Samuel Sledzieski, and Bonnie Berger. Learning the language of protein-protein interactions.Nature Communications, 17:1199, 2026. doi: 10.1038/ s41467-025-67971-3. URLhttps://doi.org/10.1038/s41467-025-67971-3
-
[28]
4M: Massively multimodal masked modeling
David Mizrahi, Roman Bachmann, O ˘guzhan Fatih Kar, Teresa Yeo, Mingfei Gao, Afshin De- hghan, and Amir Zamir. 4M: Massively multimodal masked modeling. InAdvances in Neu- ral Information Processing Systems (NeurIPS), volume 36, pages 58363–58408, 2023. URL https://arxiv.org/abs/2312.06647
arXiv 2023
-
[29]
Barthelemy Meynard-Piganeau, Christoph Feinauer, Martin Weigt, Aleksandra M. Walczak, and Thierry Mora. TULIP: A transformer-based unsupervised language model for interacting peptides and T cell receptors that generalizes to unseen epitopes.Proceedings of the National Academy of Sciences, 121(24):e2316401121, 2024. doi: 10.1073/pnas.2316401121. URL https:...
-
[30]
Dhuvarakesh Karthikeyan, Sarah N. Bennett, Amy G. Reynolds, Benjamin G. Vincent, and Alex Rubinsteyn. Conditional generation of real antigen-specific T cell receptor sequences. Nature Machine Intelligence, 7(9):1494–1509, 2025. doi: 10.1038/s42256-025-01096-6. URL https://doi.org/10.1038/s42256-025-01096-6
-
[31]
DeLisa, Jen-Tsan Ashley Chi, Ray Truant, Hector C
Leo Tianlai Chen, Zachary Quinn, Madeleine Dumas, Christina Peng, Lauren Hong, Moi- ses Lopez-Gonzalez, Alexander Mestre, Rio Watson, Sophia Vincoff, Lin Zhao, Jianli Wu, Audrey Stavrand, Mayumi Schaepers-Cheu, Tian Zi Wang, Divya Srijay, Connor Monticello, Pranay Vure, Rishab Pulugurta, Sarah Pertsemlidis, Kseniia Kholina, Shrey Goel, Matthew P. DeLisa, ...
-
[32]
Sarah M. Burbach and Bryan Briney. Improving antibody language models with native pairing. Patterns, 5(5):100967, 2024. doi: 10.1016/j.patter.2024.100967. URLhttps://doi.org/ 10.1016/j.patter.2024.100967
-
[33]
Lamb, Adalberto Claudio Quiros, Alexandrina Pancheva, Crispin J
Dan Liu, Francesca Young, Kieran D. Lamb, Adalberto Claudio Quiros, Alexandrina Pancheva, Crispin J. Miller, Craig Macdonald, David L. Robertson, and Ke Yuan. PLM- interact: extending protein language models to predict protein-protein interactions.Nature Communications, 16(1):9012, 2025. doi: 10.1038/s41467-025-64512-w. URLhttps: //doi.org/10.1038/s41467-...
-
[34]
Umberto Lupo, Damiano Sgarbossa, and Anne-Florence Bitbol. Pairing interacting pro- tein sequences using masked language modeling.Proceedings of the National Academy of Sciences, 121(27):e2311887121, 2024. doi: 10.1073/pnas.2311887121. URLhttps: //doi.org/10.1073/pnas.2311887121
-
[35]
Matthew I. J. Raybould, Alexander Greenshields-Watson, Parth Agarwal, Broncio Aguilar- Sanjuan, Tobias H. Olsen, Oliver M. Turnbull, Nele P. Quast, and Charlotte M. Deane. The observed T cell receptor space database enables paired-chain repertoire mining, coherence analysis, and language modeling.Cell Reports, 43(9):114704, 2024. doi: 10.1016/j.celrep. 20...
-
[36]
Representation Learning with Contrastive Predictive Coding
Aaron van den Oord, Yazhe Li, and Oriol Vinyals. Representation learning with contrastive predictive coding.arXiv preprint arXiv:1807.03748, 2018. doi: 10.48550/arXiv.1807.03748. URLhttps://arxiv.org/abs/1807.03748
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1807.03748 2018
-
[37]
Ralph Allan Bradley and Milton E. Terry. Rank analysis of incomplete block designs: I. The method of paired comparisons.Biometrika, 39(3/4):324–345, 1952. doi: 10.2307/2334029. URLhttps://doi.org/10.2307/2334029
-
[38]
Learning to rank using gradient descent
Chris Burges, Tal Shaked, Erin Renshaw, Ari Lazier, Matt Deeds, Nicole Hamilton, and Greg Hullender. Learning to rank using gradient descent. InProceedings of the 22nd International Conference on Machine Learning (ICML), pages 89–96. Association for Computing Machin- ery, 2005. doi: 10.1145/1102351.1102363. URLhttps://doi.org/10.1145/1102351. 1102363
-
[39]
Direct Preference Optimization: Your Language Model is Secretly a Reward Model
Rafael Rafailov, Archit Sharma, Eric Mitchell, Christopher D. Manning, Stefano Ermon, and Chelsea Finn. Direct preference optimization: Your language model is secretly a reward model. InAdvances in Neural Information Processing Systems, volume 36, pages 53728–53741, 2023. doi: 10.52202/075280-2338. URLhttps://arxiv.org/abs/2305.18290
work page internal anchor Pith review Pith/arXiv arXiv doi:10.52202/075280-2338 2023
-
[40]
Learning Transferable Visual Models From Natural Language Supervision
Alec Radford, Jong Wook Kim, Chris Hallacy, Aditya Ramesh, Gabriel Goh, Sandhini Agar- wal, Girish Sastry, Amanda Askell, Pamela Mishkin, Jack Clark, Gretchen Krueger, and Ilya Sutskever. Learning transferable visual models from natural language supervision. In Proceedings of the 38th International Conference on Machine Learning (ICML), volume 139 ofProce...
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2103.00020 2021
-
[41]
Hao Tan and Mohit Bansal. Lxmert: Learning cross-modality encoder representations from transformers. InProceedings of the 2019 conference on empirical methods in natural lan- guage processing and the 9th international joint conference on natural language process- ing (EMNLP-IJCNLP), pages 5100–5111, 2019. doi: 10.18653/v1/D19-1514. URLhttps: //arxiv.org/a...
-
[42]
Juan Jose Garau-Luis, Patrick Bordes, Liam Gonzalez, Masa Roller, Bernardo P. de Almeida, Lorenz Hexemer, Christopher Blum, Stefan Laurent, Jan Grzegorzewski, Maren Lang, Thomas Pierrot, and Guillaume Richard. Multi-modal transfer learning between biological foundation models.Advances in Neural Information Processing Systems, 37:78431–78450, 2024. doi: 10...
-
[43]
Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. ChemBERTa: Large-scale self- supervised pretraining for molecular property prediction.arXiv preprint arXiv:2010.09885,
arXiv 2010
-
[44]
doi: 10.48550/arXiv.2010.09885. URLhttps://arxiv.org/abs/2010.09885. 13
-
[45]
Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation
Mario Krenn, Florian H ¨ase, AkshatKumar Nigam, Pascal Friederich, and Alan Aspuru-Guzik. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Machine Learning: Science and Technology, 1(4):045024, 2020. doi: 10.1088/2632-2153/ aba947. URLhttps://doi.org/10.1088/2632-2153/aba947
-
[46]
Tiqing Liu, Yuhmei Lin, Xin Wen, Robert N. Jorissen, and Michael K. Gilson. Bind- ingDB in 2024: a FAIR knowledgebase of protein-small molecule binding data.Nucleic Acids Research, 53(D1):D1633–D1644, 2025. doi: 10.1093/nar/gkae1075. URLhttps: //doi.org/10.1093/nar/gkae1075
-
[47]
Mindy I. Davis, Jeremy P. Hunt, Soren Herrgard, Pietro Ciceri, Lisa M. Wodicka, Gabriel Pallares, Michael Hocker, Daniel K. Treiber, and Patrick P. Zarrinkar. Comprehensive analysis of kinase inhibitor selectivity.Nature Biotechnology, 29(11):1046–1051, 2011. doi: 10.1038/ nbt.1990. URLhttps://doi.org/10.1038/nbt.1990
-
[48]
BioSNAP datasets: Stanford biomedical network dataset collection.https://snap.stanford.edu/biodata/,
Marinka Zitnik, Rok Sosic, Sagar Maheshwari, and Jure Leskovec. BioSNAP datasets: Stanford biomedical network dataset collection.https://snap.stanford.edu/biodata/,
-
[49]
URLhttps://snap.stanford.edu/biodata/
-
[50]
Matthew A. Coelho, Magdalena E. Strauss, Alex Watterson, Sarah Cooper, Shriram Bhosle, Giuditta Illuzzi, Emre Karakoc, Cansu Dinc ¸er, Sara F. Vieira, Mamta Sharma, Marie Moullet, Daniela Conticelli, Jonas Koeppel, Katrina McCarten, Chiara M. Cattaneo, Vivien Veninga, Gabriele Picco, Leopold Parts, Josep V . Forment, Emile E. V oest, John C. Marioni, An- ...
-
[51]
Younggwang Kim, Hyeong-Cheol Oh, Seungho Lee, and Hyongbum Henry Kim. Saturation profiling of drug-resistant genetic variants using prime editing.Nature Biotechnology, 43(9): 1471–1484, 2025. doi: 10.1038/s41587-024-02465-z. URLhttps://doi.org/10.1038/ s41587-024-02465-z
-
[52]
Jonathan Frazer, Pascal Notin, Mafalda Dias, Aidan Gomez, Joseph K. Min, Kevin Brock, Yarin Gal, and Debora S. Marks. Disease variant prediction with deep generative models of evolutionary data.Nature, 599(7883):91–95, 2021. doi: 10.1038/s41586-021-04043-8. URL https://doi.org/10.1038/s41586-021-04043-8
-
[53]
Gomez, Debora Marks, and Yarin Gal
Pascal Notin, Mafalda Dias, Jonathan Frazer, Javier Marchena-Hurtado, Aidan N. Gomez, Debora Marks, and Yarin Gal. Tranception: Protein fitness prediction with autoregressive transformers and inference-time retrieval. InProceedings of the 39th International Conference on Machine Learning (ICML), volume 162 ofProceedings of Machine Learning Research, pages...
-
[54]
Pattinson, Cornelia L
Amitava Banerjee, David J. Pattinson, Cornelia L. Wincek, Paul Bunk, Armend Axhemi, Sarah R. Chapin, Saket Navlakha, and Hannah V . Meyer. T cell receptor cross-reactivity pre- diction improved by a comprehensive mutational scan database.Cell Systems, 16(8):101345,
-
[55]
URLhttps://doi.org/10.1016/j.cels.2025
doi: 10.1016/j.cels.2025.101345. URLhttps://doi.org/10.1016/j.cels.2025. 101345
-
[56]
Overton, Sandeep Kumar Dhanda, Sheridan Martini, Jason R
Randi Vita, Swapnil Mahajan, James A. Overton, Sandeep Kumar Dhanda, Sheridan Martini, Jason R. Cantrell, Daniel K. Wheeler, Alessandro Sette, and Bjoern Peters. The Immune Epitope Database (IEDB): 2018 update.Nucleic Acids Research, 47(D1):D339–D343, 2019. doi: 10.1093/nar/gky1006. URLhttps://doi.org/10.1093/nar/gky1006
-
[57]
Littler, Mark Gerstein, Anthony W
Yumeng Zhang, Zhikang Wang, Yunzhe Jiang, Dene R. Littler, Mark Gerstein, Anthony W. Purcell, Jamie Rossjohn, Hong-Yu Ou, and Jiangning Song. Epitope-anchored contrastive transfer learning for paired CD8+ T cell receptor–antigen recognition.Nature Machine Intelligence, 6(11):1344–1358, 2024. doi: 10.1038/s42256-024-00913-8. URLhttps: //doi.org/10.1038/s42...
-
[58]
Bjørn P. Y . Kwee, Marius Messemaker, Eric Marcus, Giacomo Oliveira, Wouter Scheper, Catherine J. Wu, Jonas Teuwen, and Ton N. Schumacher. STAPLER: Efficient learning of TCR-peptide specificity prediction from full-length TCR-peptide data.bioRxiv, page 2023.04.25.538237, 2023. doi: 10.1101/2023.04.25.538237. URLhttps://doi.org/10. 1101/2023.04.25.538237
-
[59]
Ceder Dens, Kris Laukens, Wout Bittremieux, and Pieter Meysman. The pitfalls of negative data bias for the T-cell epitope specificity challenge.Nature Machine Intelligence, 2023. doi: 10.1038/s42256-023-00727-0. URLhttps://doi.org/10.1038/s42256-023-00727-0
-
[60]
Eve Richardson, Yannick Jurriaan Maria Aarts, John A. Altin, Coos A. B. Baakman, Philip Bradley, Binbin Chen, Joakim Clifford, Manjima Dhar, Danielle Diepenbroek, Ethan Fast, Ragul Gowthaman, Jieling He, Vadim Karnaukhov, Dario F. Marzella, Pieter Meysman, Morten Nielsen, Jonas Birkelund Nilsson, Sebastian Nymann Deleuran, Farzaneh M. Parizi, Aurelien Pel...
-
[61]
Benchmarking of T cell receptor–epitope predictors with ePytope-TCR
Felix Drost, Anna Chernysheva, Mahmoud Albahah, Katharina Kocher, Kilian Schober, and Benjamin Schubert. Benchmarking of T cell receptor–epitope predictors with ePytope-TCR. Cell Genomics, 5(8):100946, 2025. doi: 10.1016/j.xgen.2025.100946. URLhttps://doi. org/10.1016/j.xgen.2025.100946
-
[62]
Tyler Borrman, Jennifer Cimons, Michael Cosiano, Michael Purcaro, Brian G. Pierce, Brian M. Baker, and Zhiping Weng. ATLAS: a database linking binding affinities with structures for wild-type and mutant TCR–pMHC complexes.Proteins: Structure, Function, and Bioinfor- matics, 85(5):908–916, 2017. doi: 10.1002/prot.25260. URLhttps://doi.org/10.1002/ prot.25260
-
[63]
Ido Springer, Nili Tickotsky, and Yoram Louzoun. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction.Frontiers in Im- munology, 12:664514, 2021. doi: 10.3389/fimmu.2021.664514. URLhttps://doi.org/ 10.3389/fimmu.2021.664514
-
[64]
Pieter Moris, Joey De Pauw, Anna Postovskaya, Sofie Gielis, Nicolas De Neuter, Wout Bit- tremieux, Benson Ogunjimi, Kris Laukens, and Pieter Meysman. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image clas- sification.Briefings in Bioinformatics, 22(4):bbaa318, 2021. doi: 10.1093/bib/bbaa318. URL http...
-
[65]
Yu Zhang, Xingxing Jian, Linfeng Xu, Jingjing Zhao, Manman Lu, Yong Lin, and Lu Xie. iTCep: a deep learning framework for identification of T cell epitopes by harnessing fusion features.Frontiers in Genetics, 14:1141535, 2023. doi: 10.3389/fgene.2023.1141535. URL https://doi.org/10.3389/fgene.2023.1141535
-
[66]
Xingang Peng, Yipin Lei, Peiyuan Feng, Lemei Jia, Jianzhu Ma, Dan Zhao, and Jianyang Zeng. Characterizing the interaction conformation between T-cell receptors and epitopes with deep learning.Nature Machine Intelligence, 5(4):395–407, 2023. doi: 10.1038/ s42256-023-00634-4. URLhttps://doi.org/10.1038/s42256-023-00634-4
-
[67]
T-Scan: a genome-wide method for the systematic discovery of T cell epitopes.Cell, 178(4):1016–1028,
Tomasz Kula, Mohammad H Dezfulian, Charlotte I Wang, Nouran S Abdelfattah, Zachary C Hartman, Kai W Wucherpfennig, Herbert Kim Lyerly, and Stephen J Elledge. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes.Cell, 178(4):1016–1028,
-
[68]
URLhttps://doi.org/10.1016/j.cell.2019
doi: 10.1016/j.cell.2019.07.009. URLhttps://doi.org/10.1016/j.cell.2019. 07.009
-
[69]
Ragul Gowthaman and Brian G. Pierce. TCR3d: The T cell receptor structural repertoire database.Bioinformatics, 35(24):5323–5325, 2019. doi: 10.1093/bioinformatics/btz517. URL https://doi.org/10.1093/bioinformatics/btz517. 15
-
[70]
Valerie Lin, Melyssa Cheung, Ragul Gowthaman, Maya Eisenberg, Brian M Baker, and Brian G Pierce. TCR3d 2.0: expanding the T cell receptor structure database with new structures, tools and interactions.Nucleic Acids Research, 53(D1):D604–D608, 2025. doi: 10.1093/nar/gkae840. URLhttps://doi.org/10.1093/nar/gkae840
-
[71]
Jan W Gratama, Joost WJ van Esser, Cor HJ Lamers, Claire Tournay, Bob Lowenberg, Rein- der LH Bolhuis, and Jan J Cornelissen. Tetramer-based quantification of cytomegalovirus (CMV)–specific CD8+ T lymphocytes in T-cell–depleted stem cell grafts and after transplan- tation may identify patients at risk for progressive CMV infection.Blood, The Journal of th...
-
[72]
Rafael Josip Peni ´c, Tin Vla ˇsi´c, Roland G Huber, Yue Wan, and Mile ˇSiki´c. Rinalmo: General-purpose rna language models can generalize well on structure prediction tasks.Na- ture Communications, 16(1):5671, 2025. doi: 10.1038/s41467-025-60872-5. URLhttps: //doi.org/10.1038/s41467-025-60872-5
-
[73]
Yanrong Ji, Zhihan Zhou, Han Liu, and Ramana V Davuluri. DNABERT: pre-trained bidi- rectional encoder representations from transformers model for DNA-language in genome. Bioinformatics, 37(15):2112–2120, 2021. doi: 10.1093/bioinformatics/btab083. URLhttps: //doi.org/10.1093/bioinformatics/btab083
-
[74]
ˇZiga Avsec, Vikram Agarwal, Daniel Visentin, Joseph R Ledsam, Agnieszka Grabska- Barwinska, Kyle R Taylor, Yannis Assael, John Jumper, Pushmeet Kohli, and David R Kel- ley. Effective gene expression prediction from sequence by integrating long-range interac- tions.Nature methods, 18(10):1196–1203, 2021. doi: 10.1038/s41592-021-01252-x. URL https://doi.or...
-
[75]
Zijing Gao, Qiao Liu, Wanwen Zeng, Rui Jiang, and Wing Hung Wong. Epigept: a pretrained transformer-based language model for context-specific human epigenomics.Genome Biology, 25(1):1–30, 2024. doi: 10.1186/s13059-024-03449-7. URLhttps://doi.org/10.1186/ s13059-024-03449-7
-
[76]
ˇZiga Avsec, Natasha Latysheva, Jun Cheng, Guido Novati, Kyle R. Taylor, Tom Ward, Clare Bycroft, Lauren Nicolaisen, Eirini Arvaniti, Joshua Pan, Raina Thomas, Vincent Dutordoir, Matteo Perino, Soham De, Alexander Karollus, Adam Gayoso, Toby Sargeant, Anne Mot- tram, Lai Hong Wong, Pavol Drot´ar, Adam Kosiorek, Andrew Senior, Richard Tanburn, Tay- lor App...
-
[77]
Maxim Zvyagin, Alexander Brace, Kyle Hippe, Yuntian Deng, Bin Zhang, Cindy Orozco Bo- horquez, Austin Clyde, Bharat Kale, Danilo Perez-Rivera, Heng Ma, Carla M. Mann, Michael Irvin, Defne G. Ozgulbas, Natalia Vassilieva, James Gregory Pauloski, Logan Ward, Valerie Hayot-Sasson, Murali Emani, Sam Foreman, Zhen Xie, Diangen Lin, Maulik Shukla, Weili Nie, Jo...
-
[78]
Maciej Wiatrak, Ramon Vinas Torne, Maria Ntemourtsidou, Adam M. Dinan, David C. Abel- son, Divya Arora, Maria Brbic, Aaron Weimann, and Rodrigo Andres Floto. A contextualised protein language model reveals the functional syntax of bacterial evolution.bioRxiv, 2025. doi: 10.1101/2025.07.20.665723. URLhttps://doi.org/10.1101/2025.07.20.665723
-
[79]
Li, Yepeng Huang, Marissa Sumathipala, Man Qing Liang, Alberto Valdeolivas, Ashwin N
Michelle M. Li, Yepeng Huang, Marissa Sumathipala, Man Qing Liang, Alberto Valdeolivas, Ashwin N. Ananthakrishnan, Katherine Liao, Daniel Marbach, and Marinka Zitnik. Contextual AI models for single-cell protein biology.Nature Methods, 21(8):1546–1557, 2024. doi: 10.1038/s41592-024-02341-3. URLhttps://doi.org/10.1038/s41592-024-02341-3. 16
-
[80]
Yunha Hwang, Andre L. Cornman, Elizabeth H. Kellogg, Sergey Ovchinnikov, and Pe- ter R. Girguis. Genomic language model predicts protein co-regulation and function.Na- ture Communications, 15(1):2880, 2024. doi: 10.1038/s41467-024-46947-9. URLhttps: //doi.org/10.1038/s41467-024-46947-9
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.