A Defect-Free Model of Amorphous Silicon with Pristine Electronic Structure
Pith reviewed 2026-06-26 16:04 UTC · model grok-4.3
The pith
A defect-free model of amorphous silicon generated by machine-learning molecular dynamics and checked with hybrid DFT reproduces the experimental electronic bandgap.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
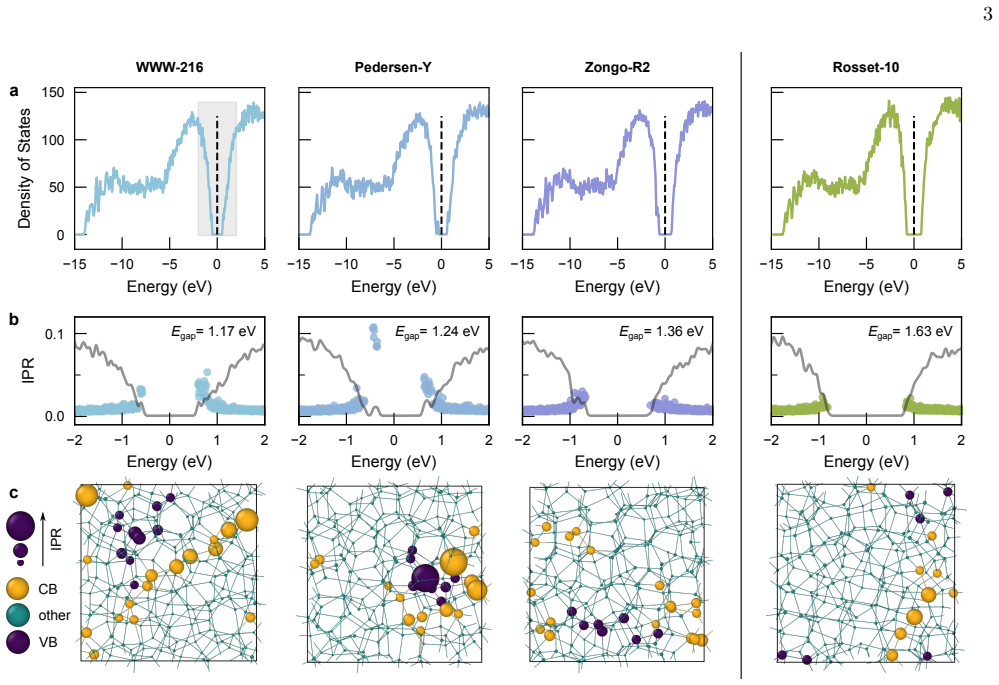
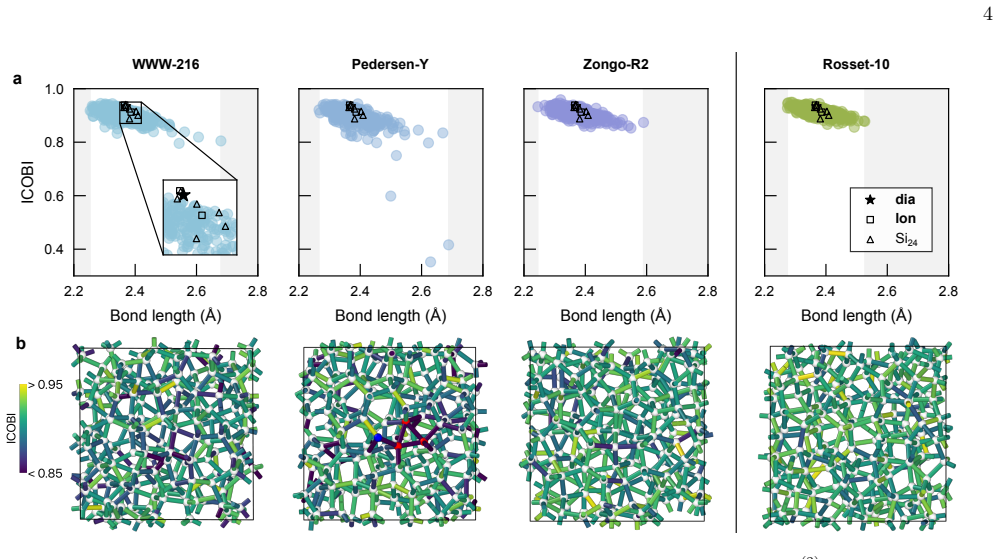
A defect-free ('ideal') model of a-Si from machine-learning-driven molecular-dynamics simulations, subsequently evaluated with hybrid-level density-functional theory computations, can accurately reproduce the experimentally observed electronic bandgap. The authors compare this model with one from the Wooten-Winer-Weaire bond-switching method and with other recent approximants to ideal a-Si, and they position the new structure as a platform for studies of band tails, optical properties, and transport.
What carries the argument
The machine-learning interatomic potential that generates the defect-free a-Si configuration during molecular dynamics, followed by independent hybrid-DFT recomputation of its electronic structure.
If this is right
- The model supplies a reference structure free of coordination defects for investigating band-tail states.
- Optical and transport calculations can be performed on the same defect-free network without artificial gap states from dangling bonds.
- Direct comparison with the WWW bond-switching model isolates the effect of generation method on electronic properties.
- The workflow can be repeated for other amorphous semiconductors to test transferability of the ML-plus-hybrid-DFT protocol.
Where Pith is reading between the lines
- If the ML potential is unbiased for electronic properties, the same structures could be used to benchmark cheaper semi-local functionals against hybrid results.
- The approach suggests that structural ideality and correct bandgap can be achieved simultaneously, which may reduce the need for post-hoc defect insertion in device modeling.
- Extension to larger cells would allow direct simulation of localized tail states and their participation in transport.
Load-bearing premise
The machine-learning potential used to create the atomic arrangement produces a configuration whose electronic bandgap, when recomputed with hybrid DFT, is not biased by the potential's training data or simulation choices.
What would settle it
A direct hybrid-DFT calculation on the ML-generated structure that yields a bandgap differing substantially from the accepted experimental value for a-Si would falsify the central claim.
Figures
read the original abstract
Amorphous silicon (a-Si) is understood to be the canonical continuous random network material, ideally defined by fully fourfold coordination. Here, we show that a defect-free ('ideal') model of a-Si from machine-learning-driven molecular-dynamics simulations [L. A. M. Rosset et al., Nat. Commun. 16, 2360 (2025)], subsequently evaluated with hybrid-level density-functional theory computations, can accurately reproduce the experimentally observed electronic bandgap. We compare this model with one resulting from the Wooten-Winer-Weaire (WWW) bond-switching approach and with other recent approximants to ideal a-Si. More broadly, our work provides a platform for studies of band tails, optical properties, and transport in a-Si.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that a defect-free ('ideal') model of amorphous silicon generated via machine-learning-driven molecular-dynamics simulations (taken from Rosset et al., Nat. Commun. 2025) reproduces the experimentally observed electronic bandgap when its electronic structure is recomputed at the hybrid-DFT level. The work compares this model to the Wooten-Winer-Weaire (WWW) bond-switching model and other recent approximants, positioning the structure as a platform for studies of band tails, optical properties, and transport.
Significance. If the central claim holds after addressing independence concerns, the result would supply a high-quality, large-scale continuous-random-network model of a-Si with a pristine gap, which is valuable for the field because traditional models often contain defects that complicate electronic-property calculations. The ML-MD route enables access to defect-free configurations at scales difficult for direct ab initio methods, and the hybrid-DFT evaluation step is a standard and appropriate choice for gap accuracy.
major comments (2)
- [Methods / Results (structure generation and electronic evaluation)] The structure originates from the ML interatomic potential of Rosset et al. (Nat. Commun. 2025). No test is reported in which an identically sized cell generated by an orthogonal protocol (WWW, ab-initio MD, or a different ML potential) is relaxed and evaluated at the same hybrid-DFT level to demonstrate that the reported bandgap match is independent of structural correlations learned during potential training (e.g., ring statistics or bond-angle distributions).
- [Abstract and Results section] The abstract states that the hybrid-DFT bandgap 'accurately reproduce[s] the experimentally observed electronic bandgap,' yet the provided text supplies neither the numerical gap value, its uncertainty, nor a direct comparison plot or table against experiment and the WWW reference. Without these quantities the quantitative claim cannot be assessed.
minor comments (2)
- [Methods] Notation for the hybrid functional (exact exchange fraction, range-separation parameters) and k-point sampling used in the DFT calculations should be stated explicitly in the methods.
- [Figures] Figure captions should include the system size (number of atoms) and the precise definition of 'defect-free' (e.g., coordination cutoff) for each model shown.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed report. The comments identify key areas where additional comparisons and quantitative clarity would strengthen the manuscript. We address each major comment below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Methods / Results (structure generation and electronic evaluation)] The structure originates from the ML interatomic potential of Rosset et al. (Nat. Commun. 2025). No test is reported in which an identically sized cell generated by an orthogonal protocol (WWW, ab-initio MD, or a different ML potential) is relaxed and evaluated at the same hybrid-DFT level to demonstrate that the reported bandgap match is independent of structural correlations learned during potential training (e.g., ring statistics or bond-angle distributions).
Authors: We agree that an explicit test with an orthogonal generation protocol evaluated at the identical hybrid-DFT level would help demonstrate that the bandgap result is not an artifact of structural features learned by the ML potential. In the revised manuscript we will add such a comparison: a WWW-generated cell of comparable size will be relaxed consistently with the ML potential and then evaluated at hybrid-DFT, with the resulting bandgap reported alongside the ML-MD result and experiment. revision: yes
-
Referee: [Abstract and Results section] The abstract states that the hybrid-DFT bandgap 'accurately reproduce[s] the experimentally observed electronic bandgap,' yet the provided text supplies neither the numerical gap value, its uncertainty, nor a direct comparison plot or table against experiment and the WWW reference. Without these quantities the quantitative claim cannot be assessed.
Authors: We acknowledge the omission. The revised manuscript will update the abstract to state the numerical hybrid-DFT bandgap value (with uncertainty) and its relation to experiment. We will also add a dedicated table or figure in the Results section that directly compares the gap values obtained for the ML-MD model, the WWW reference, and the experimental value. revision: yes
Circularity Check
No significant circularity; electronic evaluation is independent of structure generation.
full rationale
The paper's chain is: cite prior ML-MD work for a defect-free a-Si model, then recompute its electronic density of states and bandgap at hybrid DFT level, and compare to experiment and to a WWW model. No equation or claim reduces the hybrid-DFT bandgap to the ML potential's training data or loss function by construction. The hybrid-DFT step uses a different Hamiltonian and is externally falsifiable against measured gaps. Self-citation of the structure model is present but not load-bearing for the electronic result, which remains an independent computation. This is the normal case of a self-contained derivation against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
W. H. Zachariasen, J. Am. Chem. Soc.54, 3841 (1932)
1932
-
[2]
Laaziri, S
K. Laaziri, S. Kycia, S. Roorda, M. Chicoine, J. L. Robertson, J. Wang, and S. C. Moss, Phys. Rev. B60, 13520 (1999)
1999
-
[3]
Birney, J
R. Birney, J. Steinlechner, Z. Tornasi, S. MacFoy, D. Vine, A. S. Bell, D. Gibson, J. Hough, S. Rowan, P. Sortais, S. Sproules, S. Tait, I. W. Martin, and S. Reid, Phys. Rev. Lett.121, 191101 (2018)
2018
-
[4]
Wooten, K
F. Wooten, K. Winer, and D. Weaire, Phys. Rev. Lett. 54, 1392 (1985)
1985
-
[5]
Zongo, H
K. Zongo, H. Sun, C. Ouellet-Plamondon, N. Mousseau, and L. K. B´ eland, Phys. Rev. B111, 214209 (2025)
2025
-
[6]
R. H. Klazes, M. H. L. M. van den Broek, J. Bezemer, and S. Radelaar, Philos. Mag. B45, 377 (1982)
1982
-
[7]
W. Y. Ching, D. J. Lam, and C. C. Lin, Phys. Rev. B 21, 2378 (1980)
1980
-
[8]
Allan, C
G. Allan, C. Delerue, and M. Lannoo, Phys. Rev. B57, 6933 (1998)
1998
-
[9]
R. V. Meidanshahi, S. Bowden, and S. M. Goodnick, Phys. Chem. Chem. Phys.21, 13248 (2019)
2019
-
[10]
L. A. M. Rosset, D. A. Drabold, and V. L. Deringer, Nat. Commun.16, 2360 (2025)
2025
-
[11]
J. D. Morrow and V. L. Deringer, J. Chem. Phys.157, 104105 (2022)
2022
-
[12]
11 was distilled from the Si- GAP-18 potential introduced in A
The MLIP model of Ref. 11 was distilled from the Si- GAP-18 potential introduced in A. P. Bart´ ok, J. Ker- mode, N. Bernstein, and G. Cs´ anyi, Phys. Rev. X8, 041048 (2018)
2018
-
[13]
Further details are given in the Supplementary Material
-
[14]
A. P. Bart´ ok, R. Kondor, and G. Cs´ anyi, Phys. Rev. B 87, 184115 (2013)
2013
-
[15]
B. R. Djordjevi´ c, M. F. Thorpe, and F. Wooten, Phys. Rev. B52, 5685 (1995)
1995
-
[16]
Pedersen, L
A. Pedersen, L. Pizzagalli, and H. J´ onsson, New J. Phys. 19, 063018 (2017)
2017
-
[17]
Mousseau, L
N. Mousseau, L. K. B´ eland, P. Brommer, J.-F. Joly, F. El-Mellouhi, E. Machado-Charry, M.-C. Marinica, and P. Pochet, J. Phys. B: At. Mol. Phys.2012, 925278 (2012)
2012
-
[18]
P. N. Keating, Phys. Rev.145, 637 (1966)
1966
-
[19]
Y. Pan, M. Zhang, and D. Drabold, J. Non-Cryst. Solids 354, 3480 (2008)
2008
-
[20]
Opletal, T
G. Opletal, T. C. Petersen, I. K. Snook, and D. G. Mc- Culloch, J. Chem. Phys.126, 214705 (2007)
2007
-
[21]
M. J. Cliffe, M. T. Dove, D. A. Drabold, and A. L. Good- win, Phys. Rev. Lett.104, 125501 (2010)
2010
-
[22]
Pandey, P
A. Pandey, P. Biswas, and D. A. Drabold, Sci. Rep.6, 33731 (2016)
2016
-
[23]
Igram, B
D. Igram, B. Bhattarai, P. Biswas, and D. Drabold, J. Non-Cryst. Solids492, 27 (2018)
2018
-
[24]
J. Heyd, J. E. Peralta, G. E. Scuseria, and R. L. Martin, J. Chem. Phys.123, 174101 (2005)
2005
-
[25]
H. J. Xiang, B. Huang, E. Kan, S.-H. Wei, and X. G. Gong, Phys. Rev. Lett.110, 118702 (2013)
2013
-
[26]
Jarolimek, E
K. Jarolimek, E. Hazrati, R. De Groot, and G. De Wijs, Phys. Rev. Appl.8, 014026 (2017)
2017
-
[27]
J. D. Morrow, C. Ugwumadu, D. A. Drabold, S. R. Elliott, A. L. Goodwin, and V. L. Deringer, Angew. Chem. Int. Ed.63, e202403842 (2024)
2024
-
[28]
P. E. Bl¨ ochl, Phys. Rev. B50, 17953 (1994)
1994
-
[29]
Kresse and J
G. Kresse and J. Furthm¨ uller, Phys. Rev. B54, 11169 (1996)
1996
-
[30]
Kresse and D
G. Kresse and D. Joubert, Phys. Rev. B59, 1758 (1999)
1999
-
[31]
J. Heyd, G. E. Scuseria, and M. Ernzerhof, J. Chem. Phys.118, 8207 (2003)
2003
-
[32]
J. Heyd, G. E. Scuseria, and M. Ernzerhof, J. Chem. Phys.124, 219906 (2006)
2006
-
[33]
J. S. Custer, M. O. Thompson, D. C. Jacobson, J. M. Poate, S. Roorda, W. C. Sinke, and F. Spaepen, Appl. Phys. Lett.64, 437 (1994). 6
1994
-
[34]
Laaziri, S
K. Laaziri, S. Roorda, and J. M. Baribeau, J. Non- Cryst. Solids191, 193 (1995)
1995
-
[35]
Y. Liu, Y. Zhou, R. Ademuwagun, L. Walter- bos, J. George, S. R. Elliott, and V. L. Deringer, J. Am. Chem. Soc.148, 9400 (2026)
2026
-
[36]
S. C. Moss and J. F. Graczyk, Phys. Rev. Lett.23, 1167 (1969)
1969
-
[37]
L. J. Lewis, J. Non-Cryst. Solids580, 121383 (2022)
2022
-
[38]
P. M. Larsen, S. Schmidt, and J. Schiøtz, Model. Simul. Mater. Sci. Eng.24, 055007 (2016)
2016
-
[39]
J. D. Honeycutt and H. C. Andersen, J. Phys. Chem.91, 4950 (1987)
1987
-
[40]
A. S. Clarke and H. J´ onsson, Phys. Rev. E47, 3975 (1993)
1993
-
[41]
Stukowski, Model
A. Stukowski, Model. Simul. Mater. Sci. Eng.20, 045021 (2012)
2012
-
[42]
Maras, O
E. Maras, O. Trushin, A. Stukowski, T. Ala-Nissila, and H. J´ onsson, Comput. Phys. Commun.205, 13 (2016)
2016
-
[43]
Bernstein, B
N. Bernstein, B. Bhattarai, G. Cs´ anyi, D. A. Drabold, S. R. Elliott, and V. L. Deringer, Angew. Chem. Int. Ed. 58, 7057 (2019)
2019
-
[44]
Roorda and L
S. Roorda and L. J. Lewis, Science338, 1539 (2012)
2012
-
[45]
D. A. Drabold, Phys. Status Solidi Rapid Res. Lett.5, 359 (2011)
2011
-
[46]
Stukowski, Model
A. Stukowski, Model. Simul. Mater. Sci. Eng18, 015012 (2010)
2010
-
[47]
M. F. Thorpe and D. Weaire, Phys. Rev. Lett.27, 1581 (1971)
1971
-
[48]
Dong and D
J. Dong and D. A. Drabold, Phys. Rev. Lett.80, 1928 (1998)
1928
-
[49]
Urbach, Phys
F. Urbach, Phys. Rev.92, 1324 (1953)
1953
-
[50]
Aljishi, J
S. Aljishi, J. D. Cohen, S. Jin, and L. Ley, Phys. Rev. Lett.64, 2811 (1990)
1990
-
[51]
D. A. Drabold, Y. Li, B. Cai, and M. Zhang, Phys. Rev. B 83, 045201 (2011)
2011
-
[52]
D. Y. Kim, S. Stefanoski, O. O. Kurakevych, and T. A. Strobel, Nat. Mater.14, 169 (2015)
2015
-
[53]
Durandurdu, D
M. Durandurdu, D. A. Drabold, and N. Mousseau, Phys. Rev. B62, 15307 (2000)
2000
-
[54]
G. T. Barkema and N. Mousseau, Phys. Rev. B62, 4985 (2000)
2000
-
[55]
G. K. M. Thutupalli and S. G. Tomlin, J. Phys. C: Solid State Phys.10, 467 (1977)
1977
-
[56]
Park, C.-J
N.-M. Park, C.-J. Choi, T.-Y. Seong, and S.-J. Park, Phys. Rev. Lett.86, 1355 (2001)
2001
-
[57]
Huang, Y
C.-C. Huang, Y. Tang, M. van der Laan, J. van de Groep, A. F. Koenderink, and K. Dohnalov´ a, ACS Appl. Nano Mater.4, 288 (2021)
2021
-
[58]
D. A. Drabold, P. A. Fedders, O. F. Sankey, and J. D. Dow, Phys. Rev. B42, 5135 (1990)
1990
-
[59]
Prasai, P
K. Prasai, P. Biswas, and D. A. Drabold, Semi- cond. Sci. Technol.31, 073002 (2016)
2016
-
[60]
D. J. Thouless, Phys. Rep.13, 93 (1974)
1974
-
[61]
Herath, P
U. Herath, P. Tavadze, X. He, E. Bousquet, S. Singh, F. Mu˜ noz, and A. H. Romero, Comput. Phys. Commun. 251, 107080 (2020)
2020
-
[62]
L. Lang, P. Tavadze, A. Tellez, E. Bousquet, H. Xu, F. Mu˜ noz, N. Vasquez, U. Herath, and A. H. Romero, Comput. Phys. Commun.297, 109063 (2024)
2024
-
[63]
Y. Pan, F. Inam, M. Zhang, and D. A. Drabold, Phys. Rev. Lett.100, 206403 (2008)
2008
-
[64]
P. A. Fedders, D. A. Drabold, and S. Nakhmanson, Phys. Rev. B58, 15624 (1998)
1998
-
[65]
F. Inam, J. P. Lewis, and D. A. Drabold, Phys. Status Solidi A207, 599 (2010)
2010
-
[66]
Maintz, V
S. Maintz, V. L. Deringer, A. L. Tchougr´ eeff, and R. Dronskowski, J. Comput. Chem.34, 2557 (2013)
2013
-
[67]
Nelson, C
R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hau- tier, and R. Dronskowski, J. Comput. Chem.41, 1931 (2020)
1931
-
[68]
P. C. M¨ uller, C. Ertural, J. Hempelmann, and R. Dron- skowski, J. Phys. Chem. C125, 7959 (2021)
2021
-
[69]
Pauling, J
L. Pauling, J. Am. Chem. Soc.69, 542 (1947)
1947
-
[70]
Winer, I
K. Winer, I. Hirabayashi, and L. Ley, Phys. Rev. Lett. 60, 2697 (1988)
1988
-
[71]
Y. Lee, Y. Hu, D. Kim, S. Datta, and K. Cho, Phys. Rev. B105, 085201 (2022)
2022
-
[72]
H. Lin, M. Yang, X. Ru, G. Wang, S. Yin, F. Peng, C. Hong, M. Qu, J. Lu, L. Fang, C. Han, P. Procel, O. Isabella, P. Gao, Z. Li, and X. Xu, Nat. Energy8, 789 (2023)
2023
-
[73]
Oliphant, V
E. Oliphant, V. Mantena, M. Brod, G. J. Snyder, and W. Sun, Mater. Horiz.12, 3073 (2025). Supplemental Material for ‘A Defect-Free Model of Amorphous Silicon with Pristine Electronic Structure’ Louise A. M. Rosset1, Chinonso Ugwumadu2, Stephen R. Elliott3, David A. Drabold4, and V olker L. Deringer*1 1Inorganic Chemistry Laboratory, Department of Chemistr...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.