MMGNN: Multi-level, multi-color graph neural networks for molecular property prediction
Pith reviewed 2026-06-26 17:58 UTC · model grok-4.3
The pith
Molecular graphs decomposed into overlapping atom-type-pair subgraphs yield competitive property predictions on MoleculeNet benchmarks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
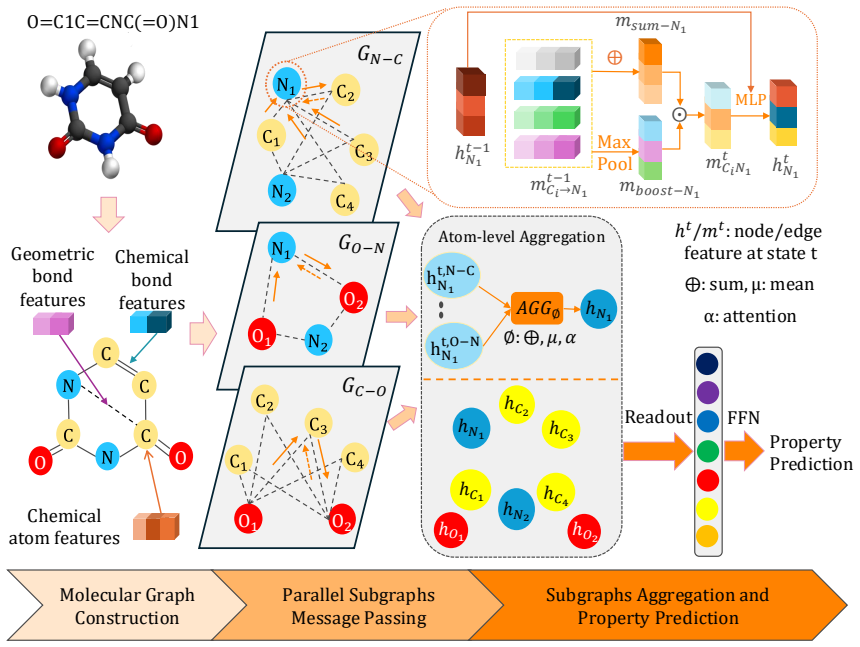
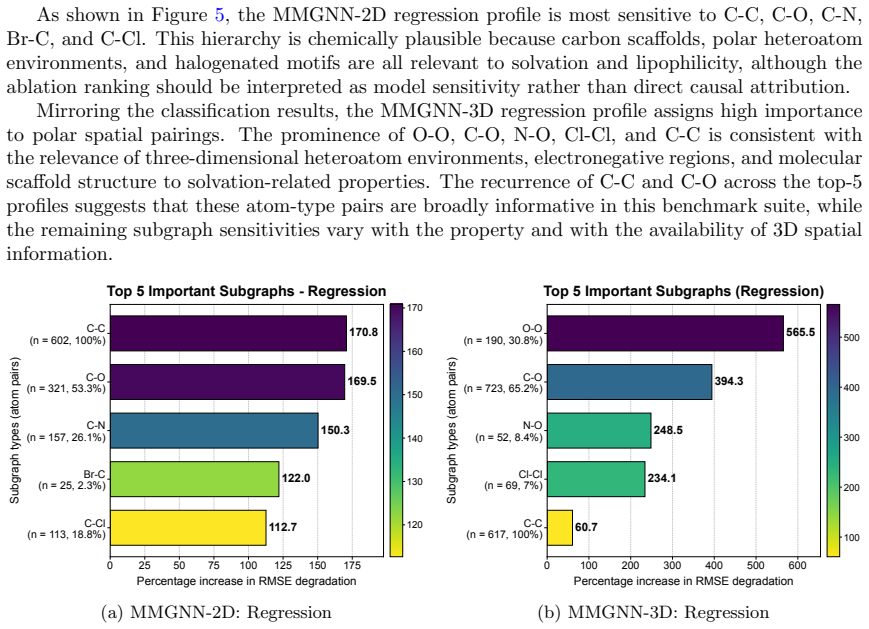
MMGNN decomposes the molecular graph into overlapping atom-type-pair-specific subgraphs, processes each with a shared communicative message-passing backbone, and aggregates atom-wise and molecule-wise representations. On five classification and three regression MoleculeNet tasks with scaffold splits, MMGNN-2D reaches macro-average AUC-ROC 0.838 and ESOL RMSE 0.803; MMGNN-3D reaches BBBP AUC-ROC 0.956 and FreeSolv RMSE 1.793. Structural analyses show how the decomposition influences learned representations.
What carries the argument
Overlapping atom-type-pair-specific subgraph decomposition, which separates interaction signals while preserving atom-level resolution before shared message passing and aggregation.
If this is right
- MMGNN-2D and MMGNN-3D show complementary strengths between topological covalent and geometric spatial representations.
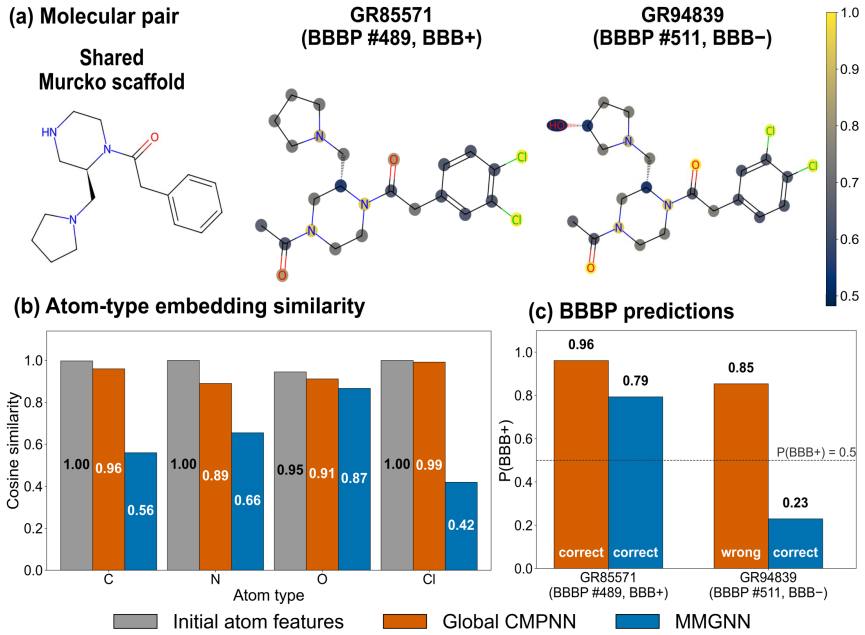
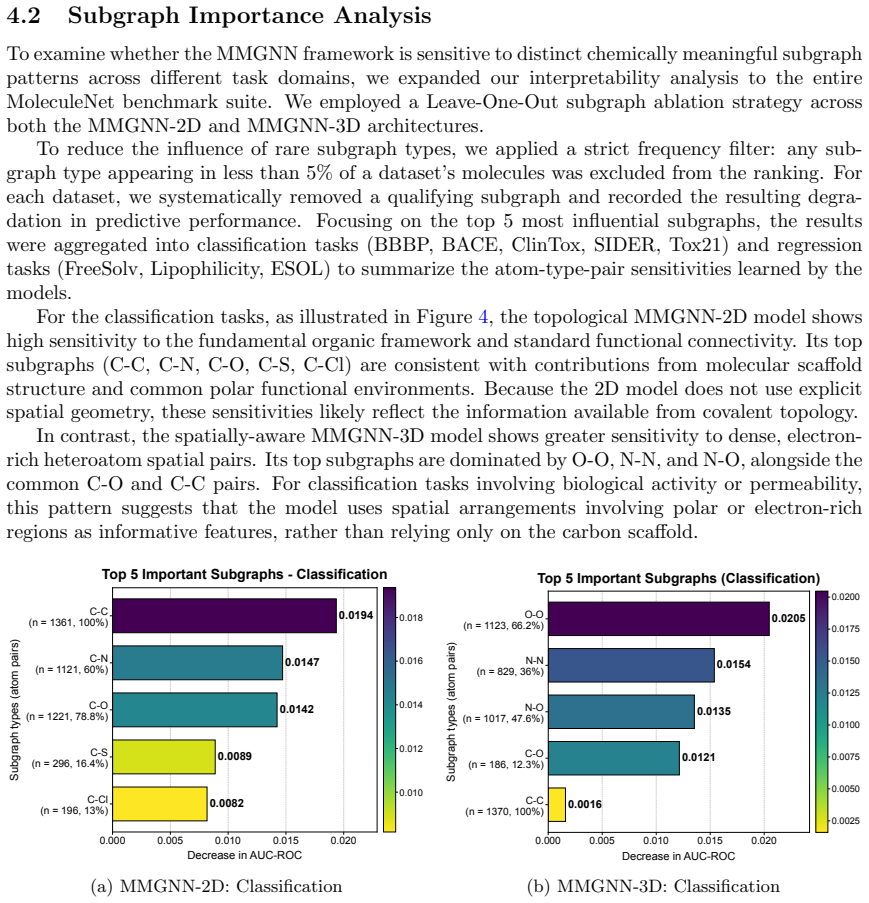
- Leave-one-out analyses reveal how the subgraph split alters atom-type-pair sensitivities in the learned representations.
- Overlapping interaction-specific graph decomposition functions as a competitive strategy compared with single-graph message passing for molecular property prediction.
Where Pith is reading between the lines
- The same decomposition idea could be tested on non-molecular graphs where node-type interactions vary, such as social or citation networks.
- It might allow shallower networks to capture effects that otherwise require many layers by structuring the input into focused subgraphs.
- Performance under random splits rather than scaffold splits would test whether the gains depend on the specific train-test separation.
- Keywords from author emphasis on multi-level and multi-color aspects suggest exploring whether the color assignment itself can be learned rather than predefined by atom types.
Load-bearing premise
That constructing separate subgraphs for each atom-type pair captures distinct interaction signals more effectively than a single unified graph while still retaining all necessary atom information.
What would settle it
A single unified graph model that matches or exceeds MMGNN performance across the same scaffold-split MoleculeNet benchmarks in repeated runs would undermine the claimed advantage of the decomposition.
Figures
read the original abstract
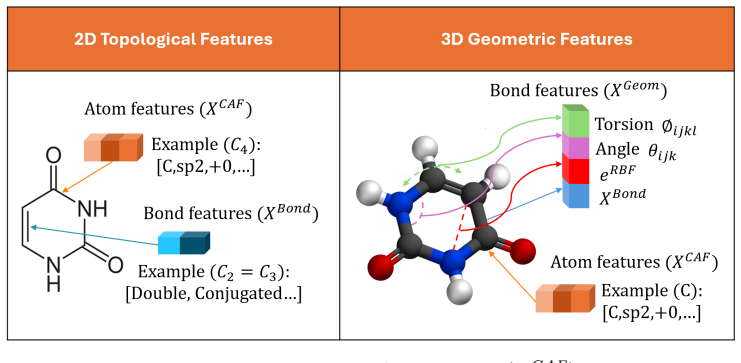
Molecular message-passing neural networks commonly propagate chemically diverse interactions through a single graph, which may mix interaction-specific signals and require deep propagation to capture long-range effects. We introduce the Multi-level, Multi-color Graph Neural Network (MMGNN), a hierarchical framework that decomposes a molecular graph into overlapping atom-type-pair-specific subgraphs while preserving atom-level resolution. MMGNN-2D constructs chemical-colored subgraphs from covalent connectivity, whereas MMGNN-3D constructs geometric-colored subgraphs from spatial proximity and augments their edges with distance, angular, and torsional descriptors. Both variants apply a shared communicative message-passing backbone to each subgraph and combine the resulting representations through atom-wise aggregation and molecular readout. We evaluated MMGNN on five classification and three regression benchmarks from MoleculeNet using common scaffold splits and five independent runs. MMGNN-2D achieved the highest macro-average AUC-ROC of 0.838 across the classification datasets and the lowest RMSE on ESOL (0.803). MMGNN-3D obtained the highest mean AUC-ROC on BBBP (0.956) and the lowest RMSE on FreeSolv (1.793), indicating complementary strengths of topological and geometric representations. Structural and leave-one-out analyses further illustrate how the subgraph decomposition affects learned representations and atom-type-pair sensitivities. These results support overlapping interaction-specific graph decomposition as a competitive strategy for molecular property prediction.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes MMGNN, a hierarchical GNN framework that decomposes a molecular graph into overlapping atom-type-pair-specific subgraphs (MMGNN-2D from covalent bonds; MMGNN-3D from spatial proximity with geometric descriptors), applies a shared communicative message-passing backbone to each subgraph, and fuses the outputs via atom-wise aggregation followed by molecular readout. On five MoleculeNet classification and three regression tasks with scaffold splits and five independent runs, MMGNN-2D reports the highest macro-average AUC-ROC (0.838) and lowest ESOL RMSE (0.803), while MMGNN-3D leads on BBBP AUC-ROC (0.956) and FreeSolv RMSE (1.793); structural and leave-one-out analyses are included to illustrate subgraph effects.
Significance. If the performance gains and ablation-style analyses hold under rigorous statistical controls, the work supplies concrete evidence that explicit interaction-specific subgraph decomposition can separate chemically diverse signals more effectively than a single unified graph while retaining atom-level resolution, offering a practical alternative to deeper message passing for molecular property prediction.
major comments (3)
- [Results] Results section (implicit in the reported macro-average AUC-ROC and RMSE values): the five-run averages are presented without error bars, standard deviations, or statistical significance tests against baselines, which directly undermines the claim that MMGNN-2D achieves the 'highest' macro-average of 0.838 and the 'lowest' ESOL RMSE of 0.803.
- [Methods] Methods / subgraph construction paragraph: the rules for selecting atom-type pairs, determining overlap, and ensuring all atom-level information is preserved in the subsequent atom-wise aggregation step are described only at high level; without explicit pseudocode or equations showing that cross-subgraph long-range dependencies are not discarded, the central assumption that decomposition captures interaction-specific signals more effectively cannot be verified.
- [Experiments] Experimental protocol: no details are supplied on hyperparameter selection, message-passing depth per subgraph, or readout functions, leaving open the possibility that reported gains arise from unstated differences in training protocol rather than the multi-color decomposition itself.
minor comments (3)
- [Abstract] The abstract and results would benefit from an explicit table listing per-dataset scores with baselines for direct comparison.
- [Introduction] Notation for 'chemical-colored' vs. 'geometric-colored' subgraphs should be formalized with a short equation or diagram to avoid ambiguity in the multi-level description.
- [Results] The leave-one-out analysis is mentioned but its quantitative impact on the main claims is not summarized in a dedicated table or figure caption.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback, which highlights important areas for improving the rigor and reproducibility of our work. We address each major comment below and will incorporate revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [Results] Results section (implicit in the reported macro-average AUC-ROC and RMSE values): the five-run averages are presented without error bars, standard deviations, or statistical significance tests against baselines, which directly undermines the claim that MMGNN-2D achieves the 'highest' macro-average of 0.838 and the 'lowest' ESOL RMSE of 0.803.
Authors: We agree that reporting only point estimates without measures of variability or statistical comparisons weakens the performance claims. In the revised manuscript, we will add standard deviations across the five independent runs to all reported metrics and include statistical significance tests (e.g., paired t-tests or Wilcoxon signed-rank tests with p-values) against the strongest baselines to substantiate the 'highest' and 'lowest' designations. revision: yes
-
Referee: [Methods] Methods / subgraph construction paragraph: the rules for selecting atom-type pairs, determining overlap, and ensuring all atom-level information is preserved in the subsequent atom-wise aggregation step are described only at high level; without explicit pseudocode or equations showing that cross-subgraph long-range dependencies are not discarded, the central assumption that decomposition captures interaction-specific signals more effectively cannot be verified.
Authors: We acknowledge that the subgraph construction details are presented at a high level. We will revise the Methods section to include explicit pseudocode for atom-type pair selection and subgraph decomposition, along with equations formalizing the overlap handling and atom-wise aggregation step. These additions will demonstrate that all atom information is retained and that cross-subgraph dependencies are not discarded by the decomposition process. revision: yes
-
Referee: [Experiments] Experimental protocol: no details are supplied on hyperparameter selection, message-passing depth per subgraph, or readout functions, leaving open the possibility that reported gains arise from unstated differences in training protocol rather than the multi-color decomposition itself.
Authors: We agree that insufficient protocol details leave room for alternative explanations of the gains. In the revised manuscript, we will add a dedicated subsection detailing the hyperparameter selection procedure (including search ranges and criteria), the message-passing depth used per subgraph, and the specific readout functions employed. We will also confirm that baselines were evaluated under equivalent training conditions to isolate the contribution of the multi-color decomposition. revision: yes
Circularity Check
No circularity: empirical architecture evaluated on public benchmarks
full rationale
The paper introduces MMGNN as a hierarchical GNN that decomposes molecular graphs into overlapping atom-type-pair subgraphs, applies message passing per subgraph, and aggregates atom-wise before readout. All reported results (AUC-ROC 0.838 macro-average, RMSE values on ESOL/FreeSolv) are direct experimental outcomes on MoleculeNet datasets using scaffold splits and multiple runs. No equations, derivations, fitted parameters renamed as predictions, or self-citation chains appear in the provided text. The central claim rests on comparative performance rather than any reduction to inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S
Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. Moleculenet: A benchmark for molecular machine learning.Chemical Science, 9(2):513–530, 2018. doi: 10.1039/C7SC02664A
-
[2]
Convolutional Networks on Graphs for Learning Molecular Fingerprints
David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Bombarell, Timothy Hirzel, Al´ an Aspuru-Guzik, and Ryan P Adams. Convolutional networks on graphs for learning molecular fingerprints.Advances in neural information processing systems, 28, 2015. doi: 10.48550/arXiv.1509.09292
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1509.09292 2015
-
[3]
Neural Message Passing for Quantum Chemistry
Justin Gilmer, Samuel S Schoenholz, Patrick F Riley, Oriol Vinyals, and George E Dahl. Neural message passing for quantum chemistry. InInternational conference on machine learning, pages 1263–1272. PMLR, 2017. doi: 10.48550/arXiv.1704.01212
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1704.01212 2017
-
[4]
How Powerful are Graph Neural Networks?
Keyulu Xu, Weihua Hu, Jure Leskovec, and Stefanie Jegelka. How powerful are graph neural networks? InInternational Conference on Learning Representations, 2019. doi: 10.48550/arXiv.1810.00826. URLhttps://openreview.net/forum?id=ryGs6iA5Km
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1810.00826 2019
-
[5]
Deeper insights into graph convolutional net- works for semi-supervised learning
Qimai Li, Zhichao Han, and Xiao-Ming Wu. Deeper insights into graph convolutional net- works for semi-supervised learning. InProceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, 2018. doi: 10.1609/aaai.v32i1.11604
-
[6]
On the bottleneck of graph neural networks and its prac- tical implications
Uri Alon and Eran Yahav. On the bottleneck of graph neural networks and its prac- tical implications. InInternational Conference on Learning Representations, 2021. doi: 10.48550/arXiv.2006.05205
-
[7]
Timothy Szocinski, Duc Duy Nguyen, and Guo-Wei Wei. Awegnn: Auto-parametrized weighted element-specific graph neural networks for molecules.Computers in Biology and Medicine, 134:104460, 2021. doi: 10.1016/j.compbiomed.2021.104460
-
[8]
Apakorn Kengkanna and Masahito Ohue. Enhancing property and activity prediction and interpretation using multiple molecular graph representations with mmgx.Communications Chemistry, 7(1), 2024. doi: 10.1038/s42004-024-01155-w
-
[9]
SchNet: A continuous-filter convolutional neural network for modeling quantum interactions
Kristof Sch¨ utt, Pieter-Jan Kindermans, Felix E Sauceda, Stefan Chmiela, Alexandre Tkatchenko, and Klaus-Robert M¨ uller. Schnet: A continuous-filter convolutional neural net- work for modeling quantum interactions. InAdvances in neural information processing systems, volume 30, 2017. doi: 10.48550/arXiv.1706.08566
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1706.08566 2017
-
[10]
E(n) equivariant graph neural networks
Victor Garcia Satorras, Emiel Hoogeboom, and Max Welling. E(n) equivariant graph neural networks. InInternational conference on machine learning, pages 9323–9332. PMLR, 2021. doi: 10.48550/arXiv.2102.09844
-
[11]
Communicative representation learning on attributed molecular graphs
Ying Song, Shuangjia Zheng, Zhangming Niu, Zhang-Hua Fu, Yefeng Yang, and Xiang-zeng Yang. Communicative representation learning on attributed molecular graphs. InProceedings of the Twenty-Ninth International Joint Conference on Artificial Intelligence, pages 2831–2838,
-
[12]
doi: 10.24963/ijcai.2020/392
-
[13]
Rdkit: Open-source cheminformatics, 2006
Greg Landrum. Rdkit: Open-source cheminformatics, 2006. URLhttps://www.rdkit.org/. 18
2006
-
[14]
Greenman, Yunsie Chung, Shih-Cheng Li, David E
Esther Heid, Kevin P Greenman, Yunsie Chung, Shih-Cheng Li, David E Graff, Florence H Vermeire, Haoyang Wu, William H Green, and Charles J McGill. Chemprop: A machine learn- ing package for chemical property prediction.Journal of Chemical Information and Modeling, 64(1):9–17, 2023. doi: 10.1021/acs.jcim.3c01250
-
[15]
Kevin Yang, Kyle Swanson, Wengong Jin, Connor Coley, Philipp Eiden, Hua Gao, Angel Guzman-Perez, Timothy Hopper, Brian Kelley, Miriam Mathea, et al. Analyzing learned molecular representations for property prediction.Journal of chemical information and mod- eling, 59(8):3370–3388, 2019. doi: 10.1021/acs.jcim.9b00237
-
[16]
Semi-Supervised Classification with Graph Convolutional Networks
Thomas N Kipf and Max Welling. Semi-supervised classification with graph convolu- tional networks. InInternational Conference on Learning Representations, 2017. doi: 10.48550/arXiv.1609.02907
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1609.02907 2017
-
[17]
Petar Veliˇ ckovi´ c, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Lio, and Yoshua Bengio. Graph attention networks. InInternational Conference on Learning Rep- resentations, 2018. doi: 10.48550/arXiv.1710.10903
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1710.10903 2018
-
[18]
Steven Kearnes, Kevin McCloskey, Marc Berndl, Vijay Pande, and Patrick Riley. Molecular graph convolutions: moving beyond fingerprints.Journal of computer-aided molecular design, 30(8):595–608, 2016. doi: 10.1007/s10822-016-9938-8
-
[19]
Zhaoping Xiong, Dingyan Wang, Xiaohong Liu, Feisheng Zhong, Xiaozhe Wan, Xutong Li, Zhaojun Li, Xiaomin Luo, Kaixian Chen, Hualiang Jiang, et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism.Journal of medicinal chemistry, 63(16):8749–8760, 2019. doi: 10.1021/acs.jmedchem.9b00959
-
[20]
Learning attributed graph representation with communicative message passing transformer
Jianwen Chen, Shuangjia Zheng, Ying Song, Jiahua Rao, and Yefeng Yang. Learning attributed graph representation with communicative message passing transformer. InProceedings of the Thirtieth International Joint Conference on Artificial Intelligence, pages 2242–2248, 2021. doi: 10.24963/ijcai.2021/309
-
[21]
Cross-dependent graph neural networks for molecular property prediction
Hehuan Ma, Yatao Bian, Yu Rong, Wenbing Huang, Tingyang Xu, Weidong Xie, Gongshen Ye, and Junzhou Huang. Cross-dependent graph neural networks for molecular property prediction. Bioinformatics, 38(7):2003–2009, 2022. doi: 10.1093/bioinformatics/btac039
-
[22]
Molecular property prediction based on graph structure learning.Bioinformatics, 40(5):btae304, 2024
Bangyi Zhao, Weixia Xu, and Jihong Guan. Molecular property prediction based on graph structure learning.Bioinformatics, 40(5):btae304, 2024. doi: 10.1093/bioinformatics/btae304. 19
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.